

TGFβ signaling plays a critical role in promoting alternative macrophage activation
- PMID: 22703233
- PMCID: PMC3406960
- DOI: 10.1186/1471-2172-13-31
TGFβ signaling plays a critical role in promoting alternative macrophage activation
Abstract
Background: Upon stimulation with different cytokines, macrophages can undergo classical or alternative activation to become M1 or M2 macrophages. Alternatively activated (or M2) macrophages are defined by their expression of specific gene products and play an important role in containing inflammation, removing apoptotic cells and repairing tissue damage. Whereas it is well-established that IL-4 can drive alternative activation, if lack of TGFβ signaling at physiological levels affects M2 polarization has not been addressed.
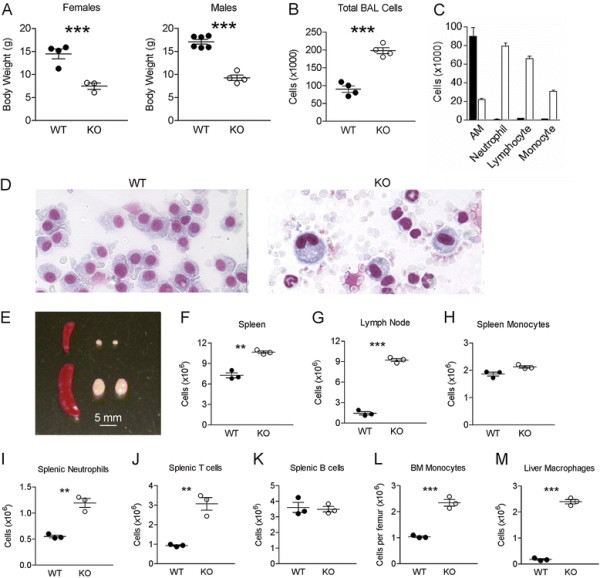
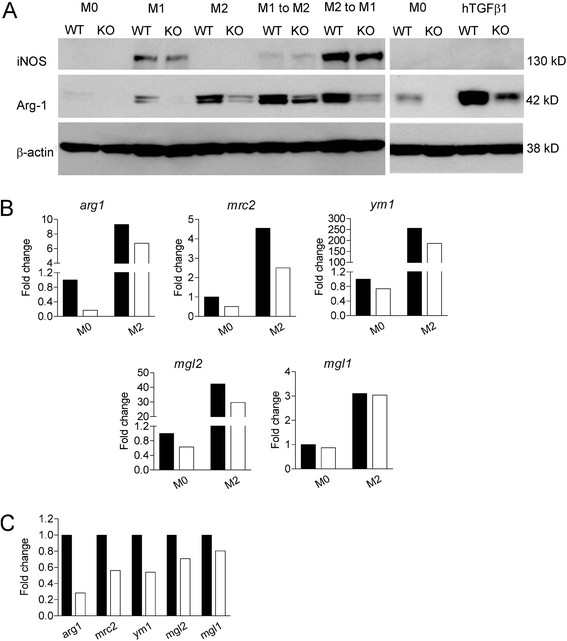
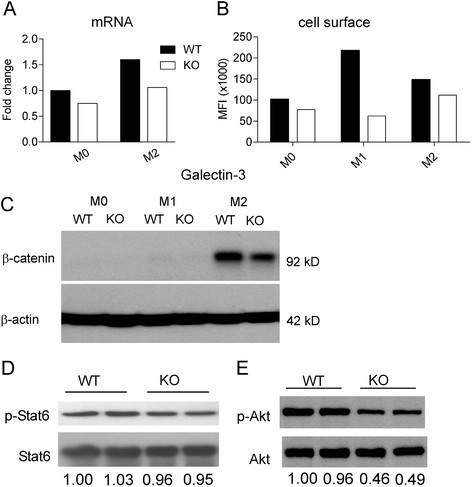
Results: Vav1-Cre x TβRIIfx/fx mice, lacking TβRII function in hematopoietic cells, exhibited uncontrolled pulmonary inflammation and developed a lethal autoimmune syndrome at young age. This was accompanied by significantly increased numbers of splenic neutrophils and T cells as well as elevated hepatic macrophage infiltration and bone marrow monocyte counts. TβRII-/- CD4+ and CD8+ T-cells in the lymph nodes and spleen expressed increased cell surface CD44, and CD69 was also higher on CD4+ lymph node T-cells. Loss of TβRII in bone marrow-derived macrophages (BMDMs) did not affect the ability of these cells to perform efferocytosis. However, these cells were defective in basal and IL-4-induced arg1 mRNA and Arginase-1 protein production. Moreover, the transcription of genes that are typically upregulated in M2-polarized macrophages, such as ym1, mcr2 and mgl2, was also decreased in peritoneal macrophages and IL-4-stimulated TβRII-/- BMDMs. We found that cell surface and mRNA expression of Galectin-3, which also regulates M2 macrophage polarization, was lower in TβRII-/- BMDMs. Very interestingly, the impaired ability of these null mutant BMDMs to differentiate into IL-4 polarized macrophages was Stat6- and Smad3-independent, but correlated with reduced levels of phospho-Akt and β-catenin.
Conclusions: Our results establish a novel biological role for TGFβ signaling in controlling expression of genes characteristic for alternatively activated macrophages. We speculate that lack of TβRII signaling reduces the anti-inflammatory M2 phenotype of macrophages because of reduced expression of these products. This would cause defects in the ability of the M2 macrophages to negatively regulate other immune cells such as T-cells in the lung, possibly explaining the systemic inflammation observed in Vav1-Cre x TβRIIfx/fx mice.
Figures
References
-
- Millan FA, Denhez F, Kondaiah P, Akhurst RJ. Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development. 1991;111:131–143. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
