

A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches
- PMID: 22705006
- PMCID: PMC3523093
- DOI: 10.1053/j.gastro.2012.06.004
A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches
Abstract
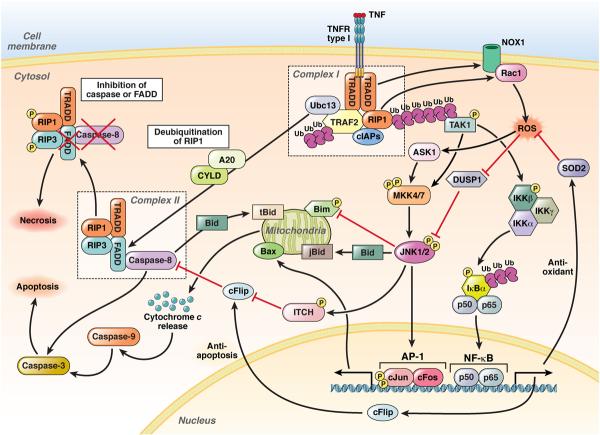
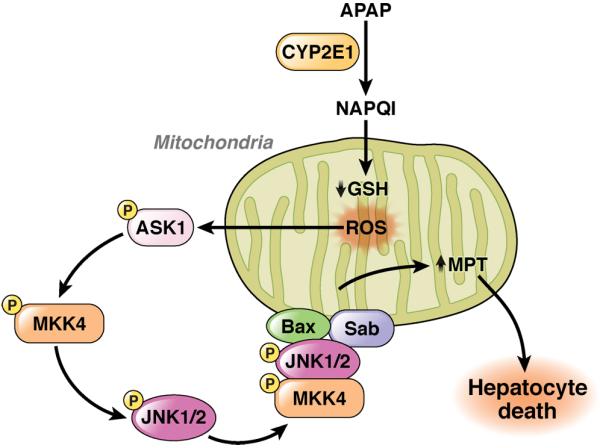
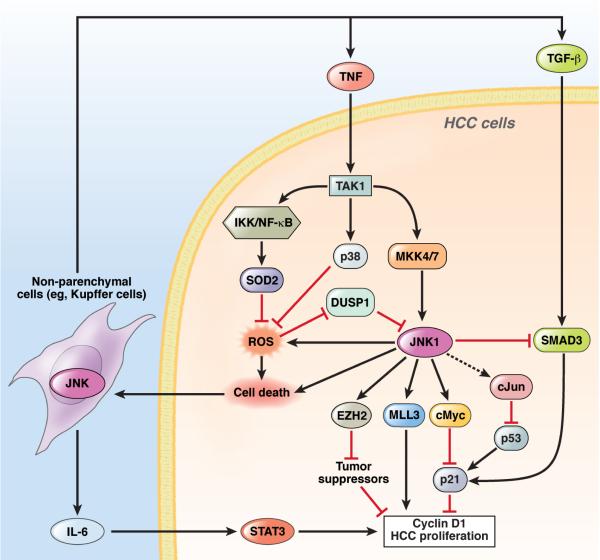
c-Jun-N-terminal kinase (JNK) is a mitogen-activated protein kinase family member that is activated by diverse stimuli, including cytokines (such as tumor necrosis factor and interleukin-1), reactive oxygen species (ROS), pathogens, toxins, drugs, endoplasmic reticulum stress, free fatty acids, and metabolic changes. Upon activation, JNK induces multiple biologic events through the transcription factor activator protein-1 and transcription-independent control of effector molecules. JNK isozymes regulate cell death and survival, differentiation, proliferation, ROS accumulation, metabolism, insulin signaling, and carcinogenesis in the liver. The biologic functions of JNK are isoform, cell type, and context dependent. Recent studies using genetically engineered mice showed that loss or hyperactivation of the JNK pathway contributes to the development of inflammation, fibrosis, cancer growth, and metabolic diseases that include obesity, hepatic steatosis, and insulin resistance. We review the functions and pathways of JNK in liver physiology and pathology and discuss findings from preclinical studies with JNK inhibitors.
Copyright © 2012 AGA Institute. Published by Elsevier Inc. All rights reserved.
Figures


References
-
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–49. - PubMed
-
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. - PubMed
-
- Kallunki T, Deng T, Hibi M, Karin M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–39. - PubMed
-
- Haeusgen W, Herdegen T, Waetzig V. The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur J Cell Biol. 2011;90:536–44. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
