

Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity
- PMID: 22706385
- PMCID: PMC3478098
- DOI: 10.1038/nm.2811
Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity
Abstract
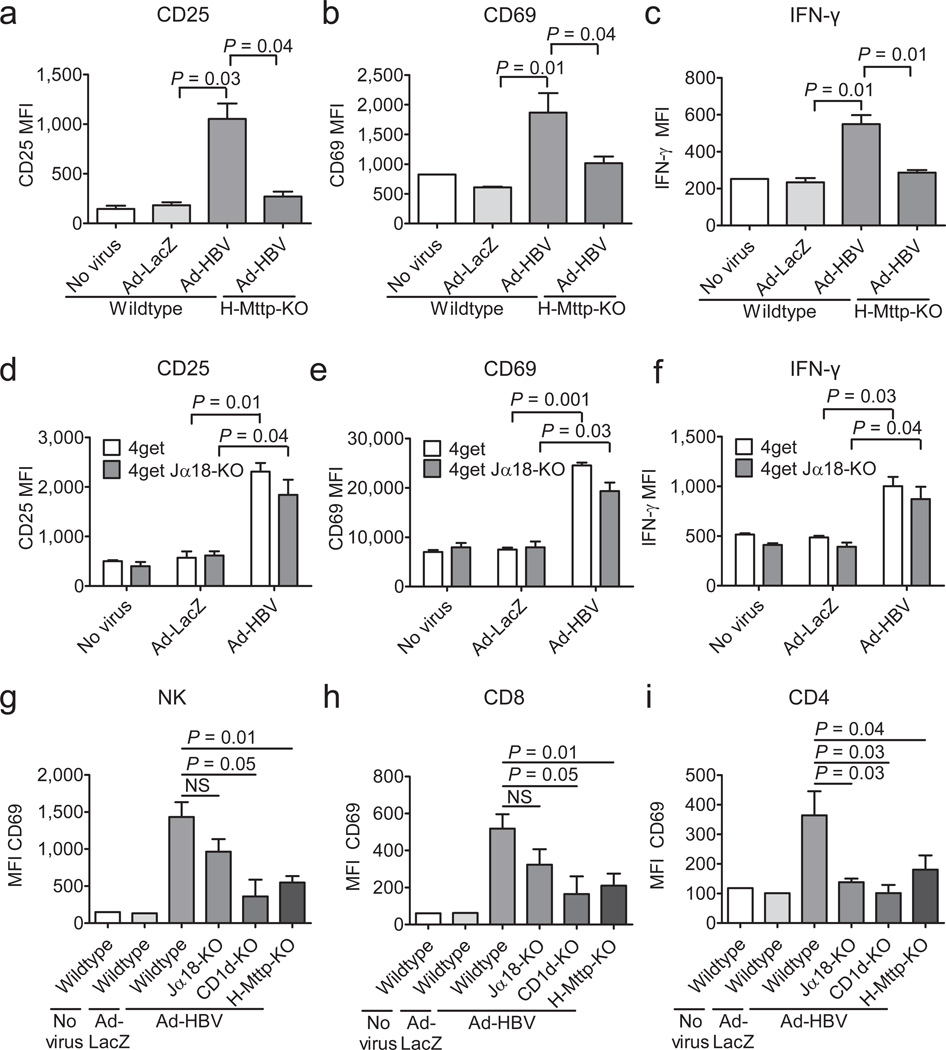
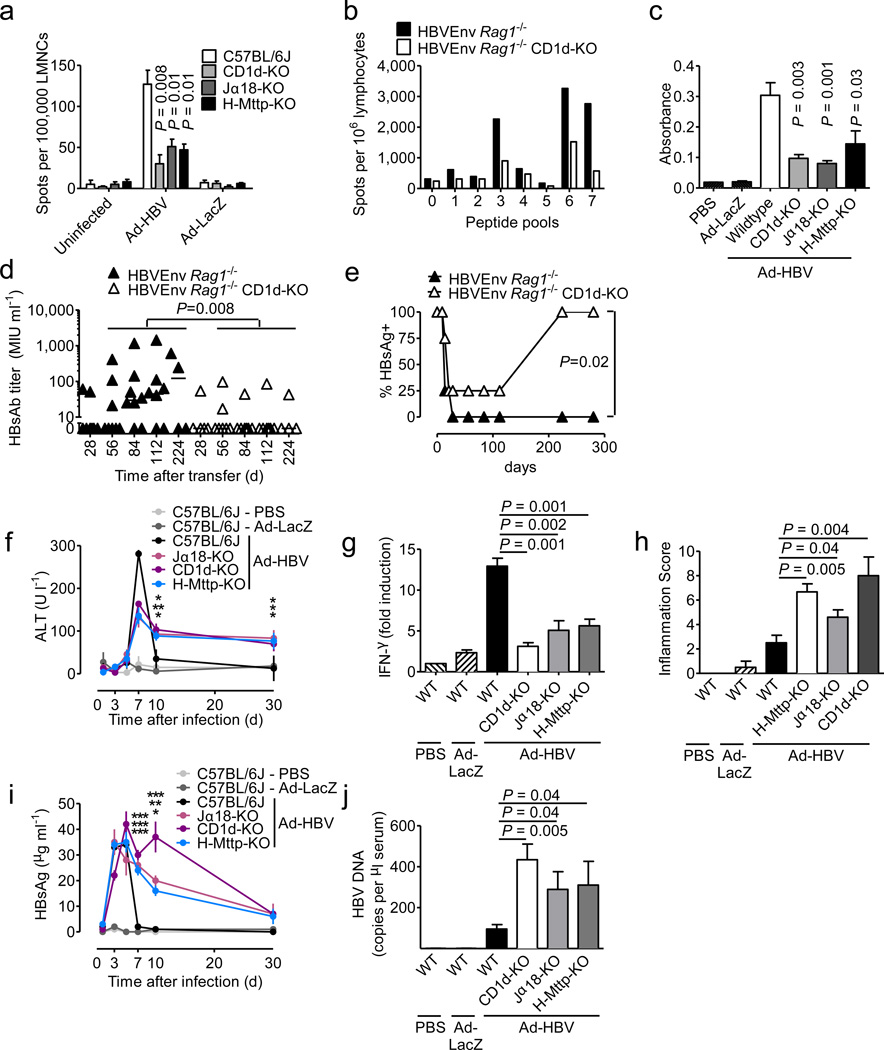
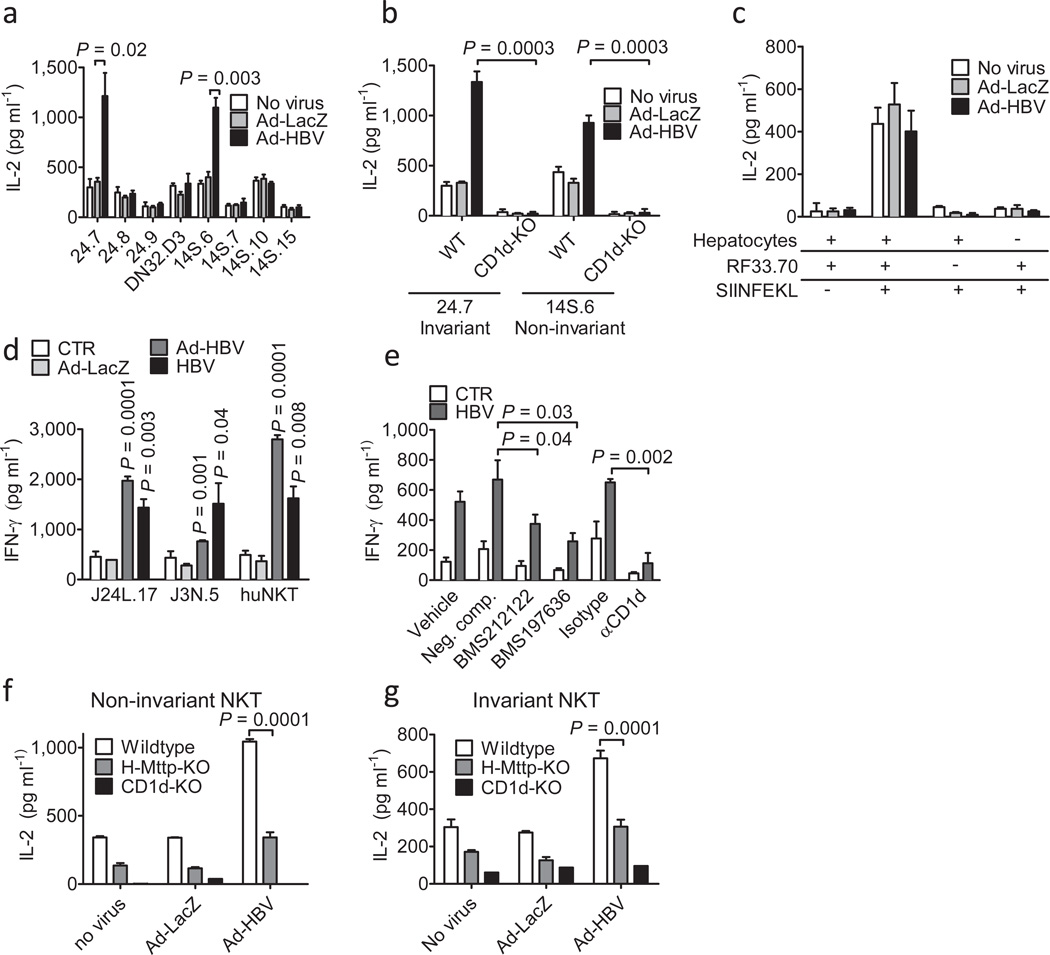
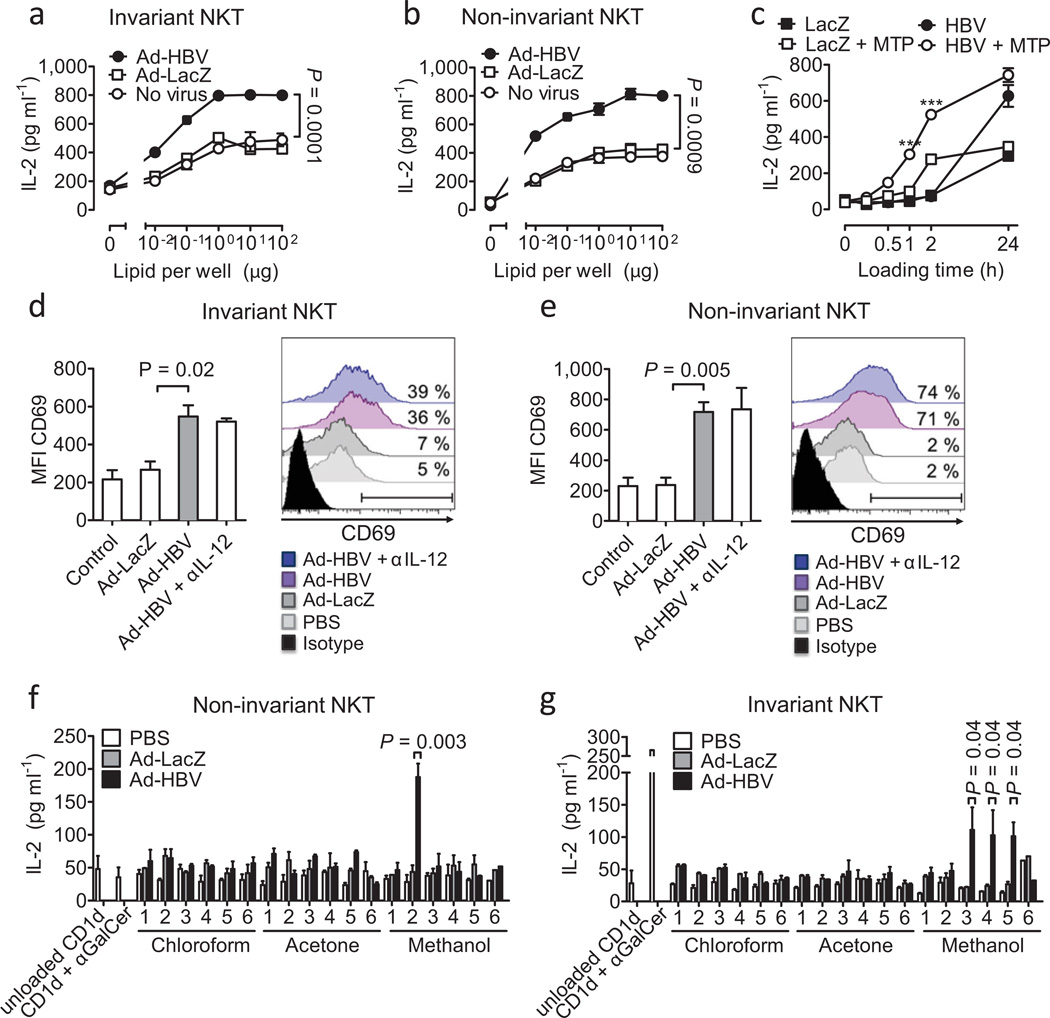
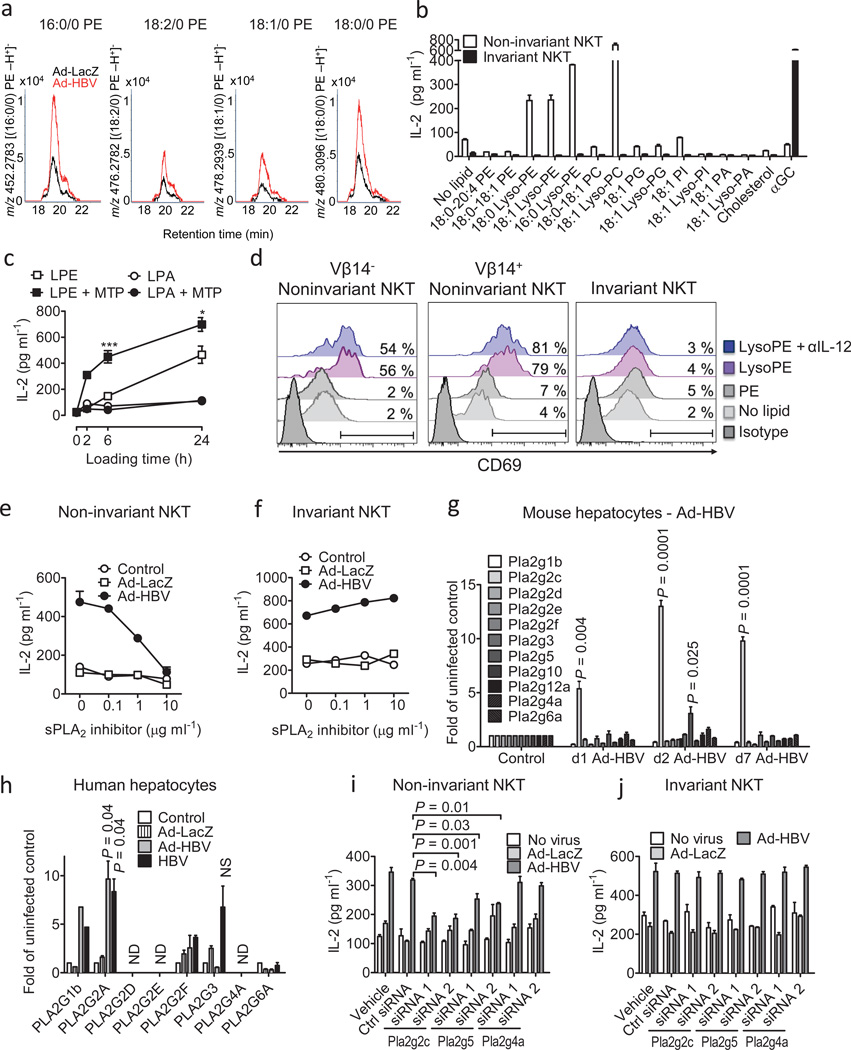
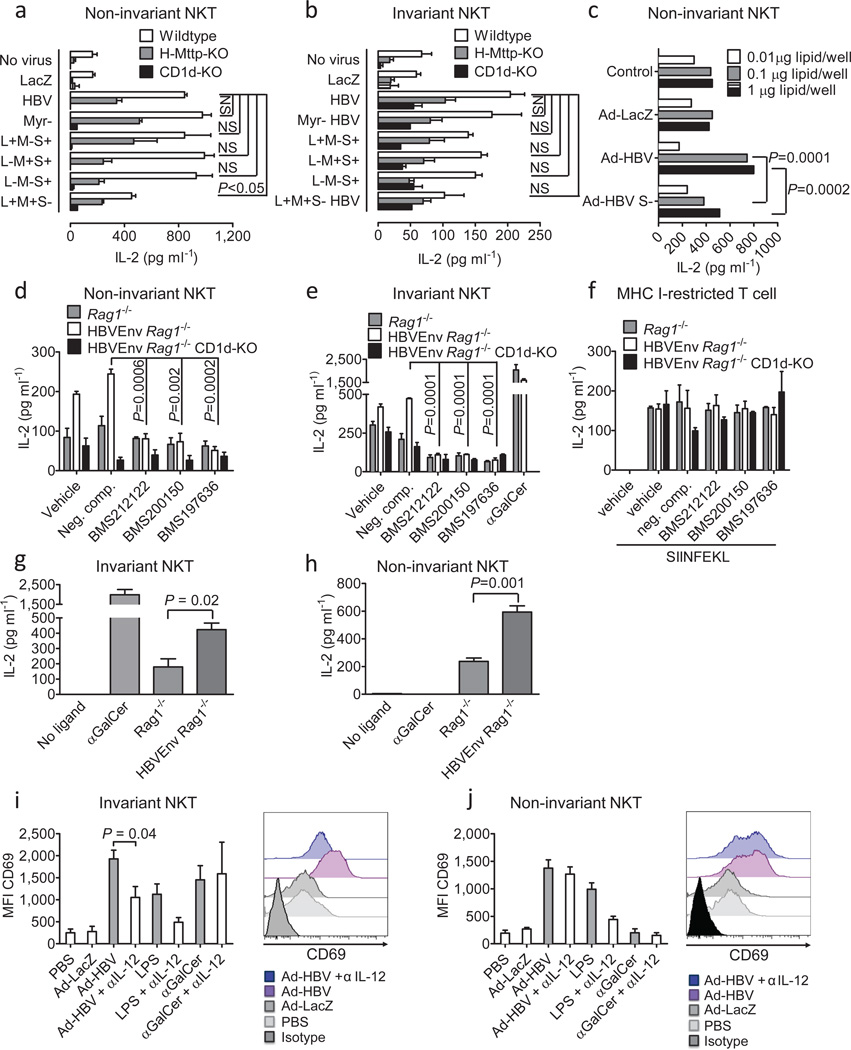
In most adult humans, hepatitis B is a self-limiting disease leading to life-long protective immunity, which is the consequence of a robust adaptive immune response occurring weeks after hepatitis B virus (HBV) infection. Notably, HBV-specific T cells can be detected shortly after infection, but the mechanisms underlying this early immune priming and its consequences for subsequent control of viral replication are poorly understood. Using primary human and mouse hepatocytes and mouse models of transgenic and adenoviral HBV expression, we show that HBV-expressing hepatocytes produce endoplasmic reticulum (ER)-associated endogenous antigenic lipids including lysophospholipids that are generated by HBV-induced secretory phospholipases and that lead to activation of natural killer T (NKT) cells. The absence of NKT cells or CD1d or a defect in ER-associated transfer of lipids onto CD1d results in diminished HBV-specific T and B cell responses and delayed viral control in mice. NKT cells may therefore contribute to control of HBV infection through sensing of HBV-induced modified self-lipids.
Figures
Comment in
-
NKT cells--an early warning system for HBV infection.Nat Med. 2012 Jul 6;18(7):1014-6. doi: 10.1038/nm.2853. Nat Med. 2012. PMID: 22772551 No abstract available.
References
-
- Yeo W, et al. Hepatitis B virus reactivation in lymphoma patients with prior resolved hepatitis B undergoing anticancer therapy with or without rituximab. J Clin Oncol. 2009;27:605–611. - PubMed
-
- Thio CL, et al. HIV-1, hepatitis B virus, and risk of liver-related mortality in the Multicenter Cohort Study (MACS) Lancet. 2002;360:1921–1926. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R37 DK044319/DK/NIDDK NIH HHS/United States
- DK51362/DK/NIDDK NIH HHS/United States
- DK44319/DK/NIDDK NIH HHS/United States
- R01 DK051362/DK/NIDDK NIH HHS/United States
- R01 AI068090/AI/NIAID NIH HHS/United States
- DK034854/DK/NIDDK NIH HHS/United States
- R01 DK046900/DK/NIDDK NIH HHS/United States
- P30 DK034854/DK/NIDDK NIH HHS/United States
- DK88199/DK/NIDDK NIH HHS/United States
- AR048632/AR/NIAMS NIH HHS/United States
- P 21530/FWF_/Austrian Science Fund FWF/Austria
- R01 DK044319/DK/NIDDK NIH HHS/United States
- DK46900/DK/NIDDK NIH HHS/United States
- AI049313/AI/NIAID NIH HHS/United States
- R01 DK088199/DK/NIDDK NIH HHS/United States
- R01 DK053056/DK/NIDDK NIH HHS/United States
- P30 DK026743/DK/NIDDK NIH HHS/United States
- AI068090/AI/NIAID NIH HHS/United States
- ZIA DK054500/ImNIH/Intramural NIH HHS/United States
- R29 DK046900/DK/NIDDK NIH HHS/United States
- R01 DK093646/DK/NIDDK NIH HHS/United States
- R56 DK046900/DK/NIDDK NIH HHS/United States
- R01 AI049313/AI/NIAID NIH HHS/United States
- R01 AR048632/AR/NIAMS NIH HHS/United States
- DK53056/DK/NIDDK NIH HHS/United States
- R56 DK053056/DK/NIDDK NIH HHS/United States
- DK026743/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
