

Constrained de novo sequencing of conotoxins
- PMID: 22709442
- PMCID: PMC3412931
- DOI: 10.1021/pr300312h
Constrained de novo sequencing of conotoxins
Abstract
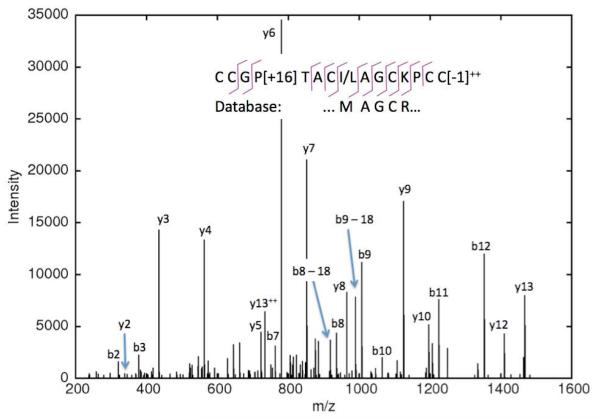
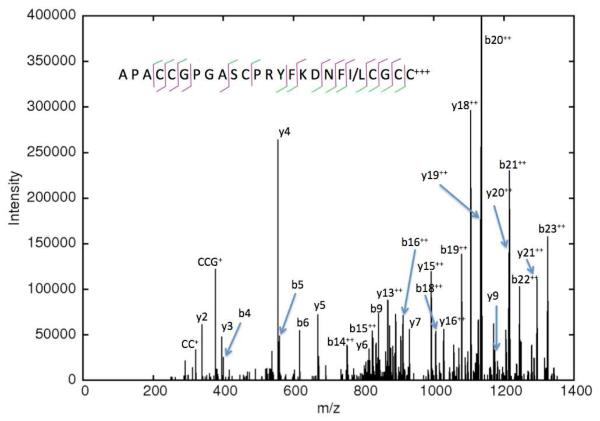
De novo peptide sequencing by mass spectrometry (MS) can determine the amino acid sequence of an unknown peptide without reference to a protein database. MS-based de novo sequencing assumes special importance in focused studies of families of biologically active peptides and proteins, such as hormones, toxins, and antibodies, for which amino acid sequences may be difficult to obtain through genomic methods. These protein families often exhibit sequence homology or characteristic amino acid content; yet, current de novo sequencing approaches do not take advantage of this prior knowledge and, hence, search an unnecessarily large space of possible sequences. Here, we describe an algorithm for de novo sequencing that incorporates sequence constraints into the core graph algorithm and thereby reduces the search space by many orders of magnitude. We demonstrate our algorithm in a study of cysteine-rich toxins from two cone snail species (Conus textile and Conus stercusmuscarum) and report 13 de novo and about 60 total toxins.
Figures
References
-
- Eng J, McCormack AL, Yates JR. An approach to correlate tandem mass spectra data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5:976–989. - PubMed
-
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. - PubMed
-
- Ma B, et al. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom. 2003;17:2337–2342. - PubMed
-
- Datta R, Bern M. Spectrum fusion: using multiple mass spectra for de novo peptide sequencing. Journal of Computational Biology. 2009;16:1–14. - PubMed
-
- Mann M, Wilm M. Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Analytical chemistry. 1994;66:4390–4399. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
