

Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration
- PMID: 22709825
- PMCID: PMC3412752
- DOI: 10.1186/1742-2094-9-130
Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration
Abstract
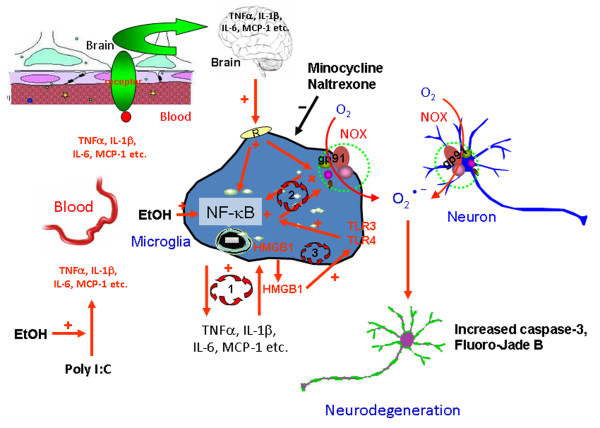
Background: Increasing evidence links systemic inflammation to neuroinflammation and neurodegeneration. We previously found that systemic endotoxin, a TLR4 agonist or TNFα, increased blood TNFα that entered the brain activating microglia and persistent neuroinflammation. Further, we found that models of ethanol binge drinking sensitized blood and brain proinflammatory responses. We hypothesized that blood cytokines contribute to the magnitude of neuroinflammation and that ethanol primes proinflammatory responses. Here, we investigate the effects of chronic ethanol on neuroinflammation and neurodegeneration triggered by toll-like receptor 3 (TLR3) agonist poly I:C.
Methods: Polyinosine-polycytidylic acid (poly I:C) was used to induce inflammatory responses when sensitized with D-galactosamine (D-GalN). Male C57BL/6 mice were treated with water or ethanol (5 g/kg/day, i.g., 10 days) or poly I:C (250 μg/kg, i.p.) alone or sequentially 24 hours after ethanol exposure. Cytokines, chemokines, microglial morphology, NADPH oxidase (NOX), reactive oxygen species (ROS), high-mobility group box 1 (HMGB1), TLR3 and cell death markers were examined using real-time PCR, ELISA, immunohistochemistry and hydroethidine histochemistry.
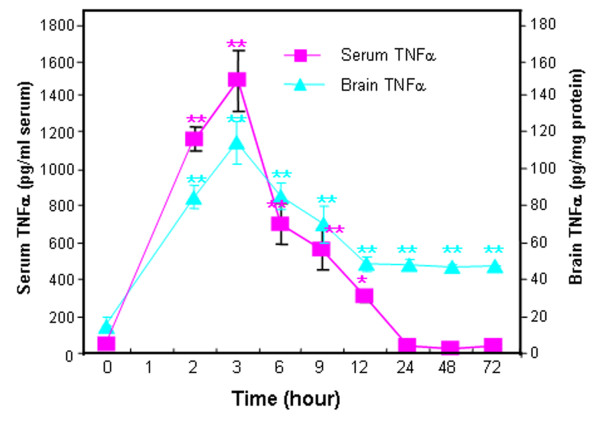
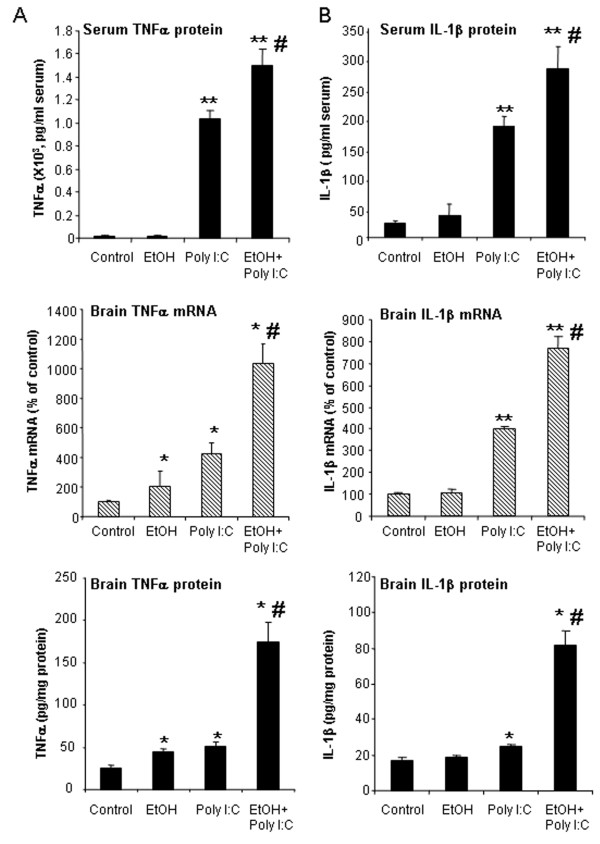
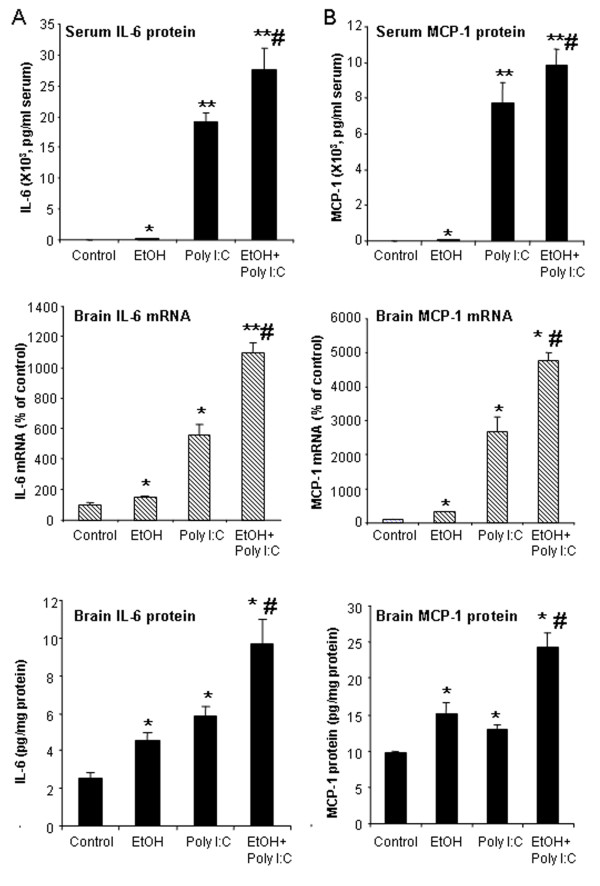
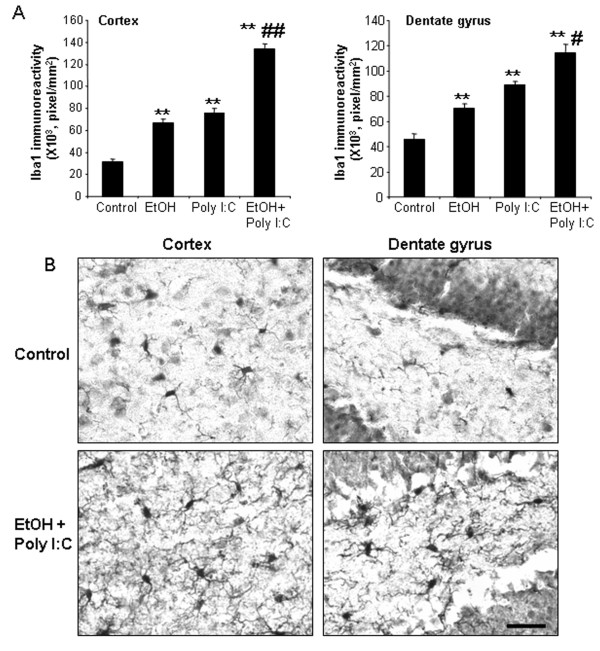
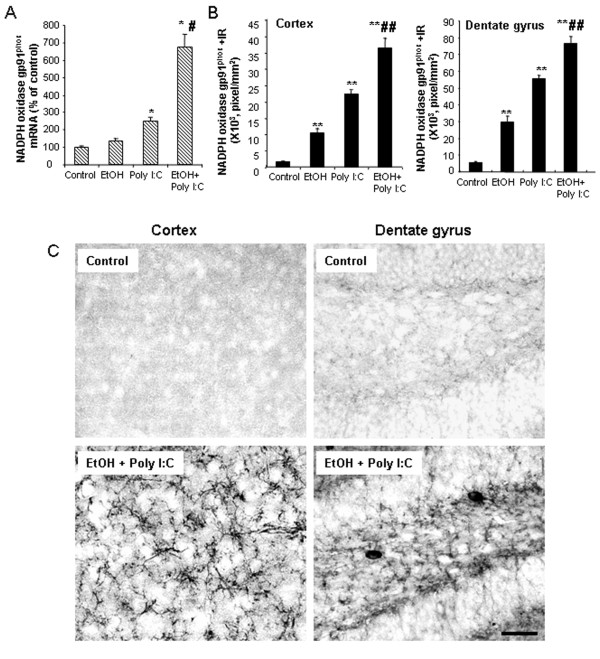
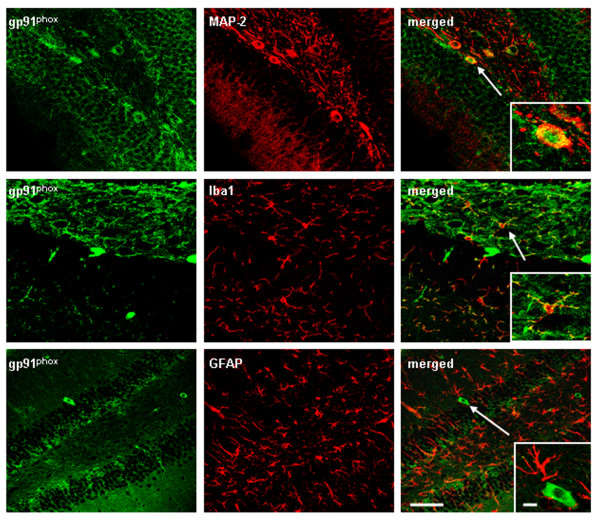
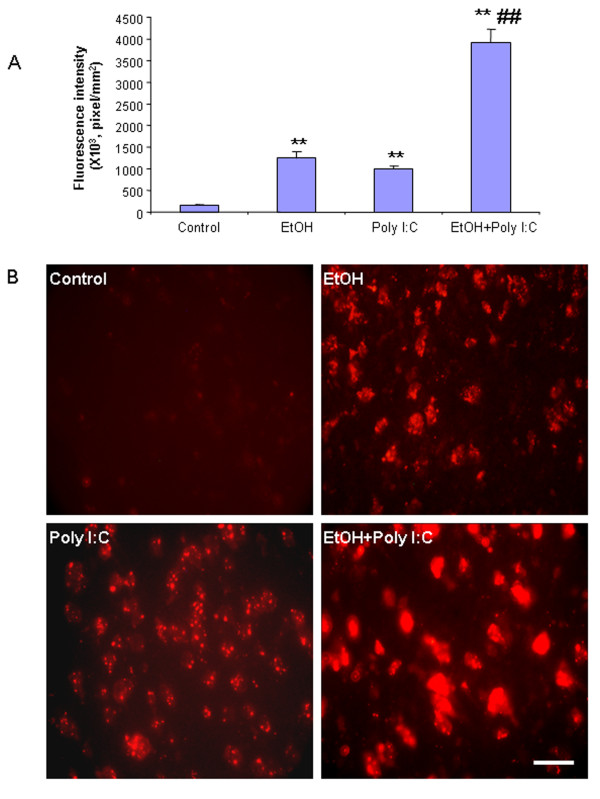
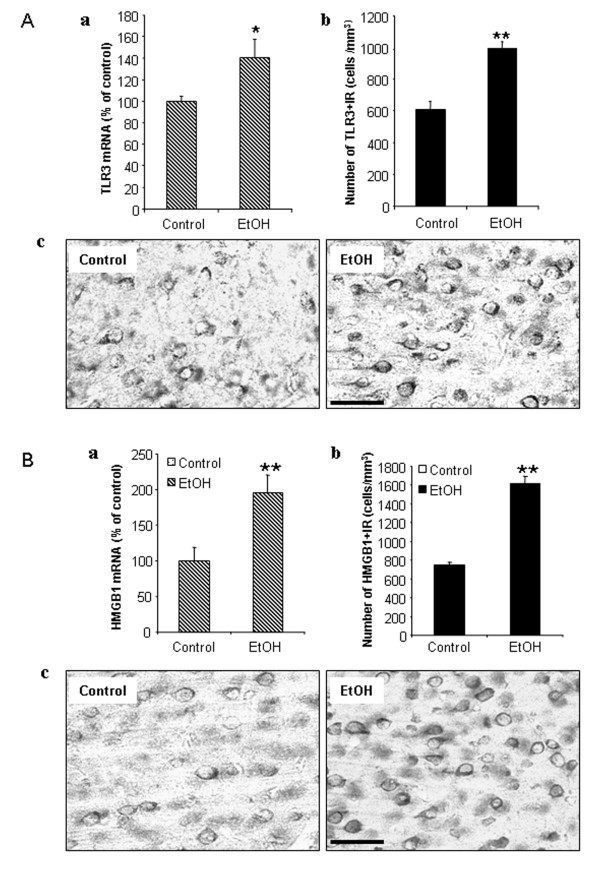
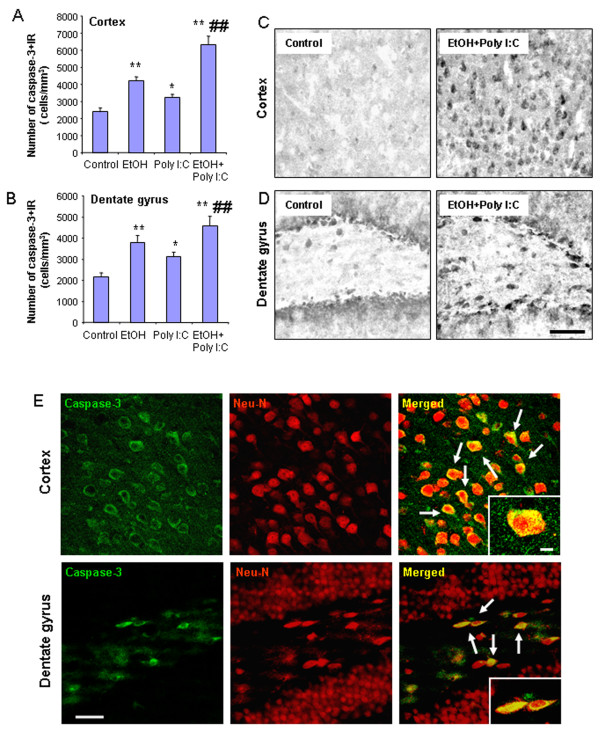
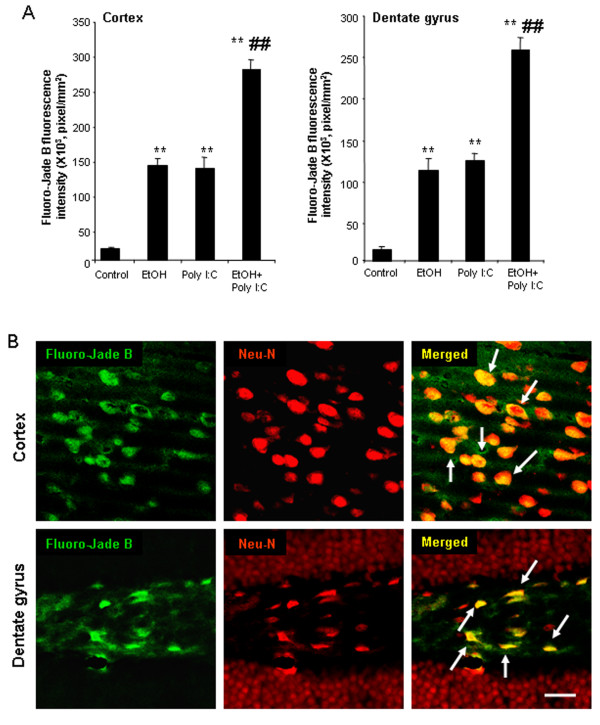
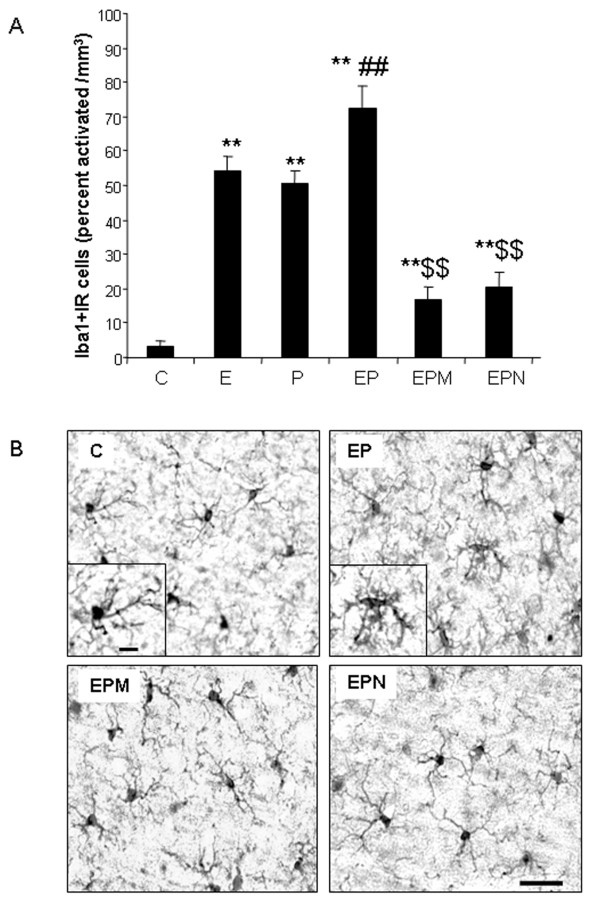
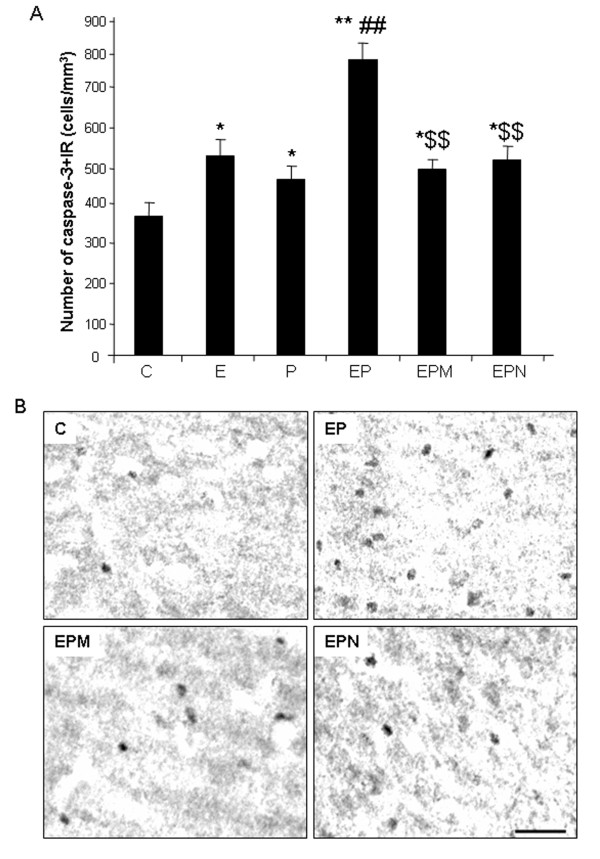
Results: Poly I:C increased blood and brain TNFα that peaked at three hours. Blood levels returned within one day, whereas brain levels remained elevated for at least three days. Escalating blood and brain proinflammatory responses were found with ethanol, poly I:C, and ethanol-poly I:C treatment. Ethanol pretreatment potentiated poly I:C-induced brain TNFα (345%), IL-1β (331%), IL-6 (255%), and MCP-1(190%). Increased levels of brain cytokines coincided with increased microglial activation, NOX gp91phox, superoxide and markers of neurodegeneration (activated caspase-3 and Fluoro-Jade B). Ethanol potentiation of poly I:C was associated with ethanol-increased expression of TLR3 and endogenous agonist HMGB1 in the brain. Minocycline and naltrexone blocked microglial activation and neurodegeneration.
Conclusions: Chronic ethanol potentiates poly I:C blood and brain proinflammatory responses. Poly I:C neuroinflammation persists after systemic responses subside. Increases in blood TNFα, IL-1β, IL-6, and MCP-1 parallel brain responses consistent with blood cytokines contributing to the magnitude of neuroinflammation. Ethanol potentiation of TLR3 agonist responses is consistent with priming microglia-monocytes and increased NOX, ROS, HMGB1-TLR3 and markers of neurodegeneration. These studies indicate that TLR3 agonists increase blood cytokines that contribute to neurodegeneration and that ethanol binge drinking potentiates these responses.
Figures
Similar articles
-
NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration.J Neuroinflammation. 2012 Jan 12;9:5. doi: 10.1186/1742-2094-9-5. J Neuroinflammation. 2012. PMID: 22240163 Free PMC article.
-
Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment.J Neuroinflammation. 2008 Mar 18;5:10. doi: 10.1186/1742-2094-5-10. J Neuroinflammation. 2008. PMID: 18348728 Free PMC article.
-
Ethanol, TLR3, and TLR4 Agonists Have Unique Innate Immune Responses in Neuron-Like SH-SY5Y and Microglia-Like BV2.Alcohol Clin Exp Res. 2017 May;41(5):939-954. doi: 10.1111/acer.13368. Epub 2017 Mar 30. Alcohol Clin Exp Res. 2017. PMID: 28273337 Free PMC article.
-
Alcohol, HMGB1, and Innate Immune Signaling in the Brain.Alcohol Res. 2024 Aug 8;44(1):04. doi: 10.35946/arcr.v44.1.04. eCollection 2024. Alcohol Res. 2024. PMID: 39135668 Free PMC article. Review.
-
Converging actions of alcohol on liver and brain immune signaling.Int Rev Neurobiol. 2014;118:359-80. doi: 10.1016/B978-0-12-801284-0.00011-7. Int Rev Neurobiol. 2014. PMID: 25175869 Review.
Cited by
-
Binge ethanol exposure in advanced age elevates neuroinflammation and early indicators of neurodegeneration and cognitive impairment in female mice.Brain Behav Immun. 2024 Feb;116:303-316. doi: 10.1016/j.bbi.2023.12.034. Epub 2023 Dec 25. Brain Behav Immun. 2024. PMID: 38151165 Free PMC article.
-
Tumour Necrosis Factor in Neuroplasticity, Neurogenesis and Alcohol Use Disorder.Brain Plast. 2020 Dec 29;6(1):47-66. doi: 10.3233/BPL-190095. Brain Plast. 2020. PMID: 33680846 Free PMC article. Review.
-
Microglia-Induced Maladaptive Plasticity Can Be Modulated by Neuropeptides In Vivo.Neural Plast. 2015;2015:135342. doi: 10.1155/2015/135342. Epub 2015 Jul 26. Neural Plast. 2015. PMID: 26273481 Free PMC article. Review.
-
Innate Immunity and Alcohol.J Clin Med. 2019 Nov 14;8(11):1981. doi: 10.3390/jcm8111981. J Clin Med. 2019. PMID: 31739600 Free PMC article. Review.
-
Healthy lifestyles and wellbeing reduce neuroinflammation and prevent neurodegenerative and psychiatric disorders.Front Neurosci. 2023 Feb 15;17:1092537. doi: 10.3389/fnins.2023.1092537. eCollection 2023. Front Neurosci. 2023. PMID: 36875655 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
