

Counteracting effects operating on Src homology 2 domain-containing protein-tyrosine phosphatase 2 (SHP2) function drive selection of the recurrent Y62D and Y63C substitutions in Noonan syndrome
- PMID: 22711529
- PMCID: PMC3411048
- DOI: 10.1074/jbc.M112.350231
Counteracting effects operating on Src homology 2 domain-containing protein-tyrosine phosphatase 2 (SHP2) function drive selection of the recurrent Y62D and Y63C substitutions in Noonan syndrome
Abstract
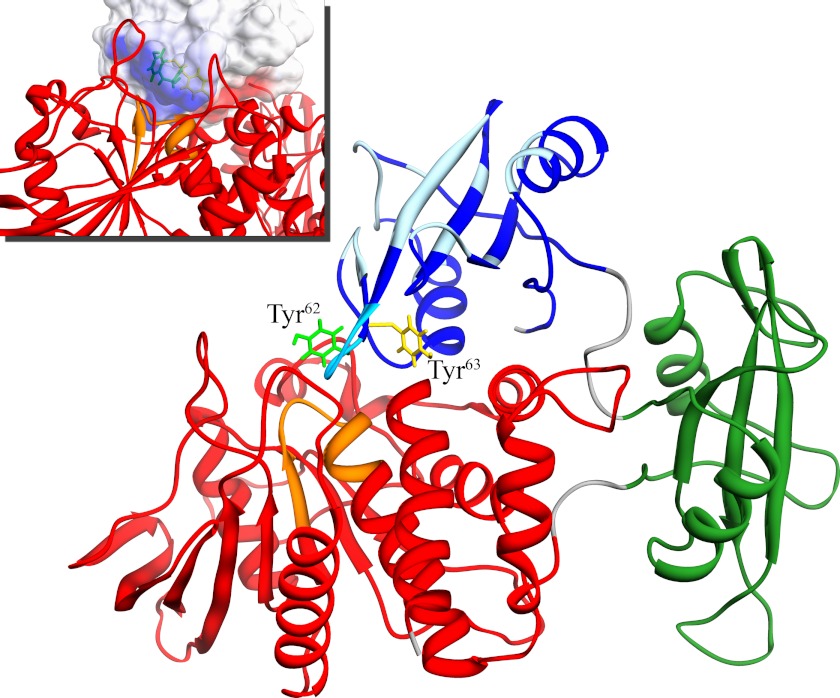
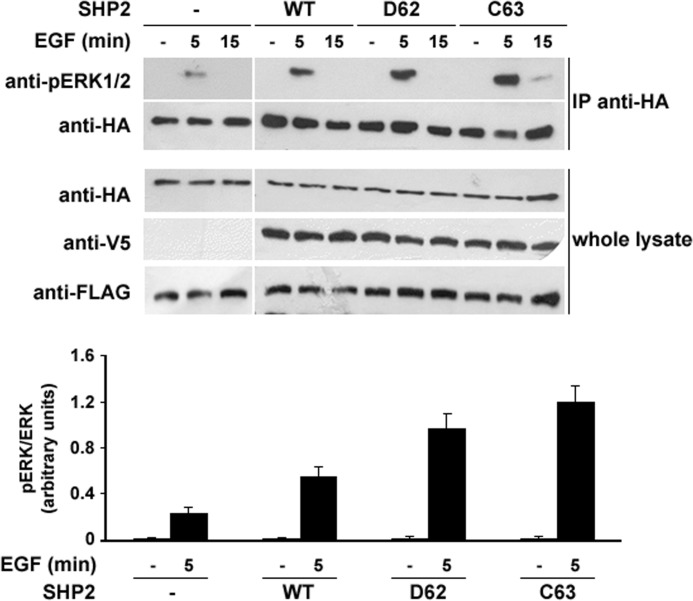
Activating mutations in PTPN11 cause Noonan syndrome, the most common nonchromosomal disorder affecting development and growth. PTPN11 encodes SHP2, an Src homology 2 (SH2) domain-containing protein-tyrosine phosphatase that positively modulates RAS function. Here, we characterized functionally all possible amino acid substitutions arising from single-base changes affecting codons 62 and 63 to explore the molecular mechanisms lying behind the largely invariant occurrence of the Y62D and Y63C substitutions recurring in Noonan syndrome. We provide structural and biochemical data indicating that the autoinhibitory interaction between the N-SH2 and protein-tyrosine phosphatase (PTP) domains is perturbed in both mutants as a result of an extensive structural rearrangement of the N-SH2 domain. Most mutations affecting Tyr(63) exerted an unpredicted disrupting effect on the structure of the N-SH2 phosphopeptide-binding cleft mediating the interaction of SHP2 with signaling partners. Among all the amino acid changes affecting that codon, the disease-causing mutation was the only substitution that perturbed the stability of the inactive conformation of SHP2 without severely impairing proper phosphopeptide binding of N-SH2. On the other hand, the disruptive effect of the Y62D change on the autoinhibited conformation of the protein was balanced, in part, by less efficient binding properties of the mutant. Overall, our data demonstrate that the selection-by-function mechanism acting as driving force for PTPN11 mutations affecting codons 62 and 63 implies balancing of counteracting effects operating on the allosteric control of the function of SHP2.
Figures





References
-
- Tartaglia M., Mehler E. L., Goldberg R., Zampino G., Brunner H. G., Kremer H., van der Burgt I., Crosby A. H., Ion A., Jeffery S., Kalidas K., Patton M. A., Kucherlapati R. S., Gelb B. D. (2001) Mutations in PTPN11, encoding the protein-tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 29, 465–468 - PubMed
-
- Allanson J. E. (2007) Noonan syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 145C, 274–279 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
