

Neuromyelitis optica: aquaporin-4 based pathogenesis mechanisms and new therapies
- PMID: 22713791
- PMCID: PMC3676174
- DOI: 10.1016/j.biocel.2012.06.013
Neuromyelitis optica: aquaporin-4 based pathogenesis mechanisms and new therapies
Abstract
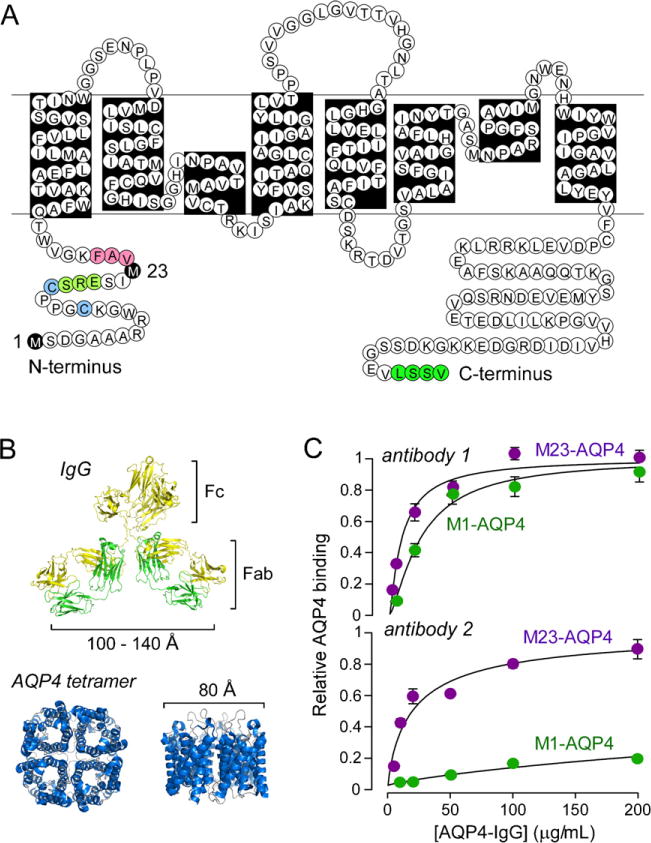
Neuromyelitis optica (NMO) is an autoimmune 'aquaporinopathy' of the central nervous system that causes inflammatory demyelinating lesions primarily in spinal cord and optic nerve, leading to paralysis and blindness. NMO lesions show loss of aquaporin-4 (AQP4), GFAP and myelin, infiltration of granulocytes and macrophages, and perivascular deposition of activated complement. Most patients with NMO are seropositive for immunoglobulin autoantibodies (AQP4-IgG) against AQP4, the principal water channel of astrocytes. There is strong evidence that AQP4-IgG is pathogenic in NMO, probably by a mechanism involving complement-dependent astrocyte cytotoxicity, causing leukocyte infiltration, cytokine release and blood-brain barrier disruption, which leads to oligodendrocyte death, myelin loss and neuron death. Here, we review the evidence for this and alternative proposed NMO pathogenesis mechanisms, such as AQP4-IgG-induced internalization of AQP4 and glutamate transporters, complement-independent cell-mediated cytotoxicity, and AQP4-IgG inhibition of AQP4 water transport function. Based on the initiating pathogenic role of AQP4-IgG binding to astrocyte AQP4 in NMO, selective blocker therapies are under development in which AQP4-targeted monoclonal antibodies or small molecules block binding of AQP4-IgG to astrocytes and consequent downstream pathology.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures






References
-
- Antoine JC, Camdessanche JP, Absi L, Lassabliere F, Feasson L. Devic disease and thymoma with anti-central nervous system and antithymus antibodies. Neurology. 2004;62:978–80. - PubMed
-
- Auguste KI, Jin S, Uchida K, Yan D, Manley GT, Papadopoulos MC, et al. Greatly impaired migration of implanted aquaporin-4-deficient astroglial cells in mouse brain toward a site of injury. FASEB Journal. 2007;21:108–16. - PubMed
-
- Balbi P, Salvini S, Fundaro C, Frazzitta G, Maestri R, Mosah D, et al. The clinical spectrum of late-onset Alexander disease: a systematic literature review. Journal of Neurology. 2010;257:1955–62. - PubMed
-
- Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. 2006;53:631–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
