

Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking
- PMID: 22716043
- PMCID: PMC3405771
- DOI: 10.1021/jm300687e
Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking
Abstract
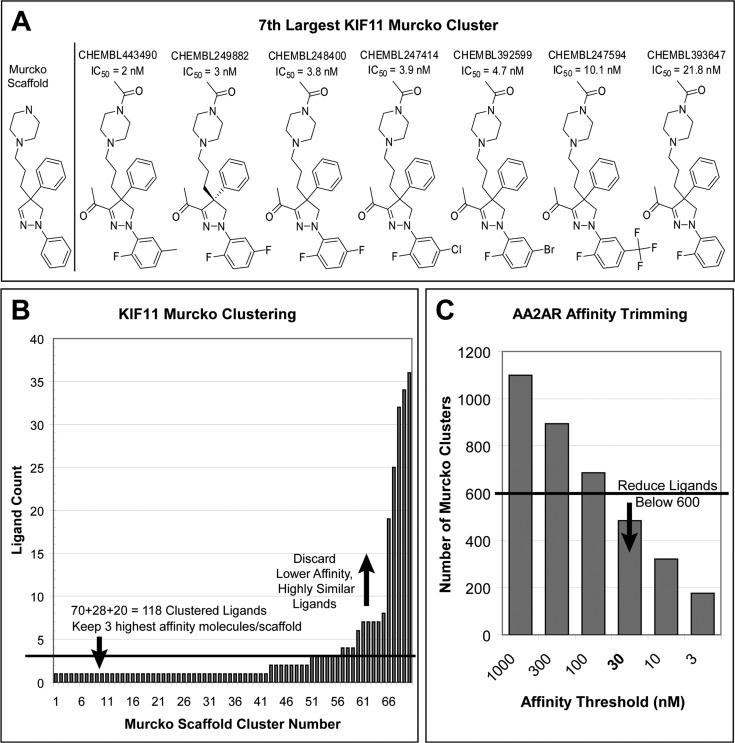
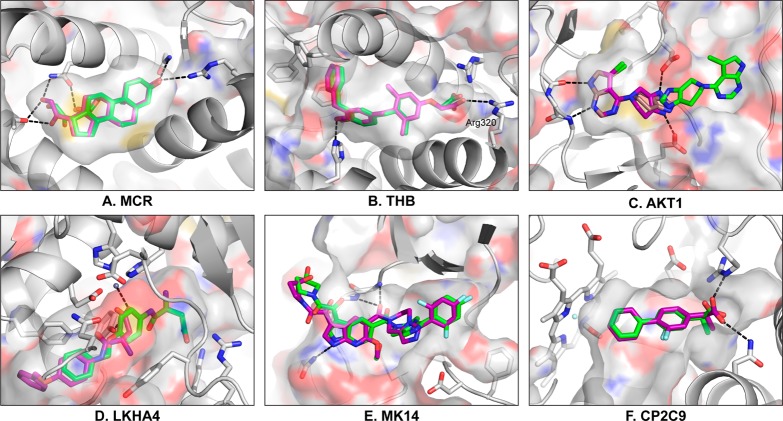
A key metric to assess molecular docking remains ligand enrichment against challenging decoys. Whereas the directory of useful decoys (DUD) has been widely used, clear areas for optimization have emerged. Here we describe an improved benchmarking set that includes more diverse targets such as GPCRs and ion channels, totaling 102 proteins with 22886 clustered ligands drawn from ChEMBL, each with 50 property-matched decoys drawn from ZINC. To ensure chemotype diversity, we cluster each target's ligands by their Bemis-Murcko atomic frameworks. We add net charge to the matched physicochemical properties and include only the most dissimilar decoys, by topology, from the ligands. An online automated tool (http://decoys.docking.org) generates these improved matched decoys for user-supplied ligands. We test this data set by docking all 102 targets, using the results to improve the balance between ligand desolvation and electrostatics in DOCK 3.6. The complete DUD-E benchmarking set is freely available at http://dude.docking.org.
Figures




Similar articles
-
Benchmarking sets for molecular docking.J Med Chem. 2006 Nov 16;49(23):6789-801. doi: 10.1021/jm0608356. J Med Chem. 2006. PMID: 17154509 Free PMC article.
-
Community benchmarks for virtual screening.J Comput Aided Mol Des. 2008 Mar-Apr;22(3-4):193-9. doi: 10.1007/s10822-008-9189-4. Epub 2008 Feb 14. J Comput Aided Mol Des. 2008. PMID: 18273555 Review.
-
Property-Unmatched Decoys in Docking Benchmarks.J Chem Inf Model. 2021 Feb 22;61(2):699-714. doi: 10.1021/acs.jcim.0c00598. Epub 2021 Jan 25. J Chem Inf Model. 2021. PMID: 33494610 Free PMC article.
-
Virtual decoy sets for molecular docking benchmarks.J Chem Inf Model. 2011 Feb 28;51(2):196-202. doi: 10.1021/ci100374f. Epub 2011 Jan 5. J Chem Inf Model. 2011. PMID: 21207928
-
Benchmarking Data Sets for the Evaluation of Virtual Ligand Screening Methods: Review and Perspectives.J Chem Inf Model. 2015 Jul 27;55(7):1297-307. doi: 10.1021/acs.jcim.5b00090. Epub 2015 Jun 18. J Chem Inf Model. 2015. PMID: 26038804 Review.
Cited by
-
Describing inhibitor specificity for the amino acid transporter LAT1 from metainference simulations.Biophys J. 2022 Dec 6;121(23):4476-4491. doi: 10.1016/j.bpj.2022.11.001. Epub 2022 Nov 11. Biophys J. 2022. PMID: 36369754 Free PMC article.
-
Homology modeling of Forkhead box protein C2: identification of potential inhibitors using ligand and structure-based virtual screening.Mol Divers. 2023 Aug;27(4):1661-1674. doi: 10.1007/s11030-022-10519-0. Epub 2022 Sep 1. Mol Divers. 2023. PMID: 36048303 Free PMC article.
-
Protease Inhibitors in View of Peptide Substrate Databases.J Chem Inf Model. 2016 Jun 27;56(6):1228-35. doi: 10.1021/acs.jcim.6b00064. Epub 2016 Jun 9. J Chem Inf Model. 2016. PMID: 27247997 Free PMC article.
-
Discovery of dual kinase inhibitors targeting VEGFR2 and FAK: structure-based pharmacophore modeling, virtual screening, and molecular docking studies.BMC Chem. 2024 Feb 12;18(1):29. doi: 10.1186/s13065-024-01130-5. BMC Chem. 2024. PMID: 38347617 Free PMC article.
-
Identification of New Potential Inhibitors of Quorum Sensing through a Specialized Multi-Level Computational Approach.Molecules. 2021 Apr 29;26(9):2600. doi: 10.3390/molecules26092600. Molecules. 2021. PMID: 33946907 Free PMC article.
References
-
- Kitchen D. B.; Decornez H.; Furr J. R.; Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Rev. Drug Discovery 2004, 3, 935–949. - PubMed
-
- Gruneberg S.; Stubbs M. T.; Klebe G. Successful virtual screening for novel inhibitors of human carbonic anhydrase: strategy and experimental confirmation. J. Med. Chem. 2002, 45, 3588–3602. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
