

Evolutionary genomics of mycovirus-related dsRNA viruses reveals cross-family horizontal gene transfer and evolution of diverse viral lineages
- PMID: 22716092
- PMCID: PMC3483285
- DOI: 10.1186/1471-2148-12-91
Evolutionary genomics of mycovirus-related dsRNA viruses reveals cross-family horizontal gene transfer and evolution of diverse viral lineages
Abstract
Background: Double-stranded (ds) RNA fungal viruses are typically isometric single-shelled particles that are classified into three families, Totiviridae, Partitiviridae and Chrysoviridae, the members of which possess monopartite, bipartite and quadripartite genomes, respectively. Recent findings revealed that mycovirus-related dsRNA viruses are more diverse than previously recognized. Although an increasing number of viral complete genomic sequences have become available, the evolution of these diverse dsRNA viruses remains to be clarified. This is particularly so since there is little evidence for horizontal gene transfer (HGT) among dsRNA viruses.
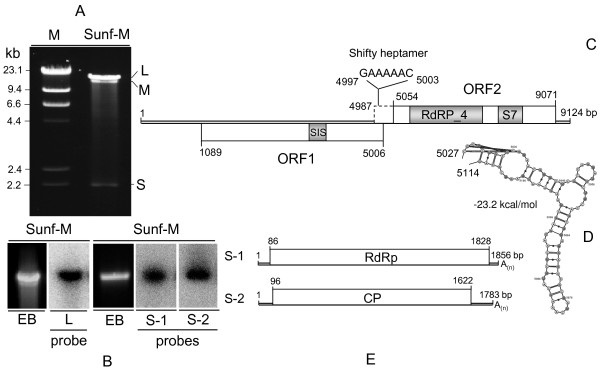
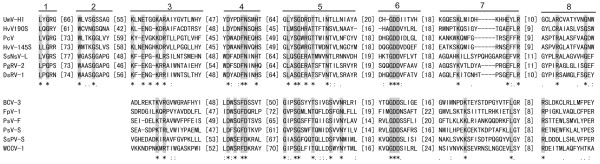
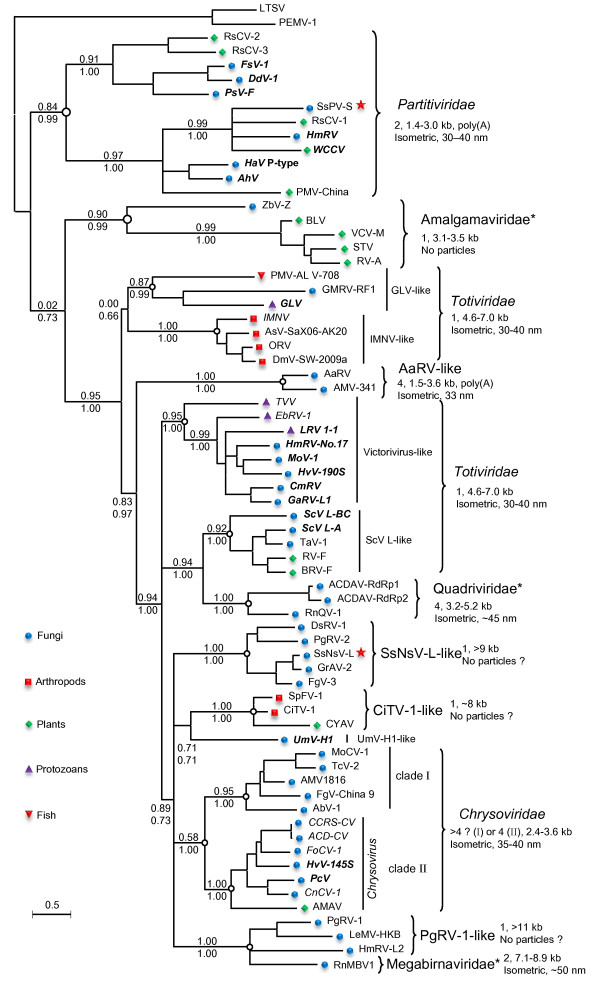
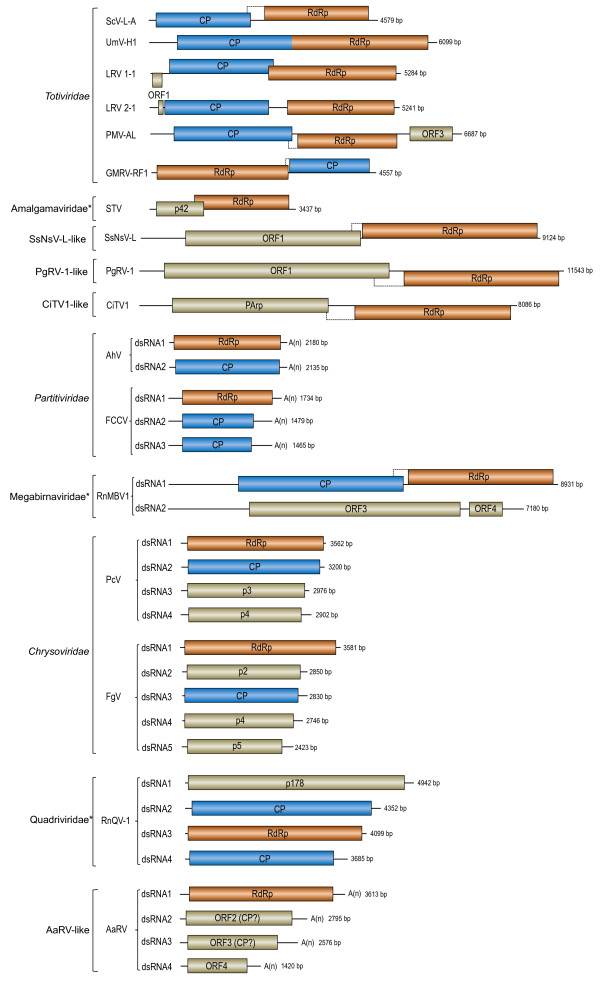
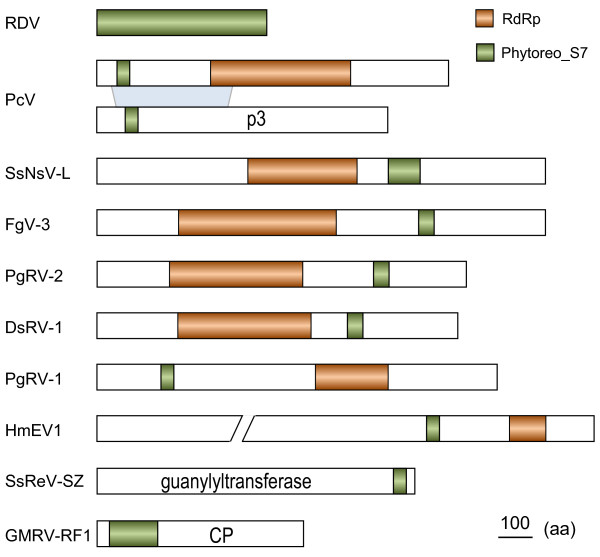
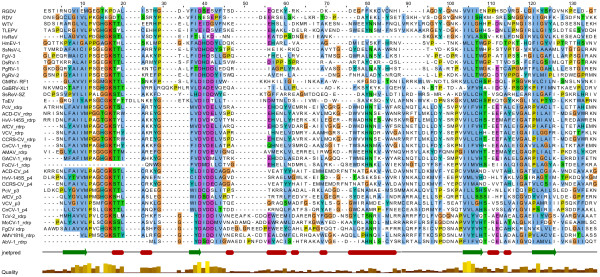
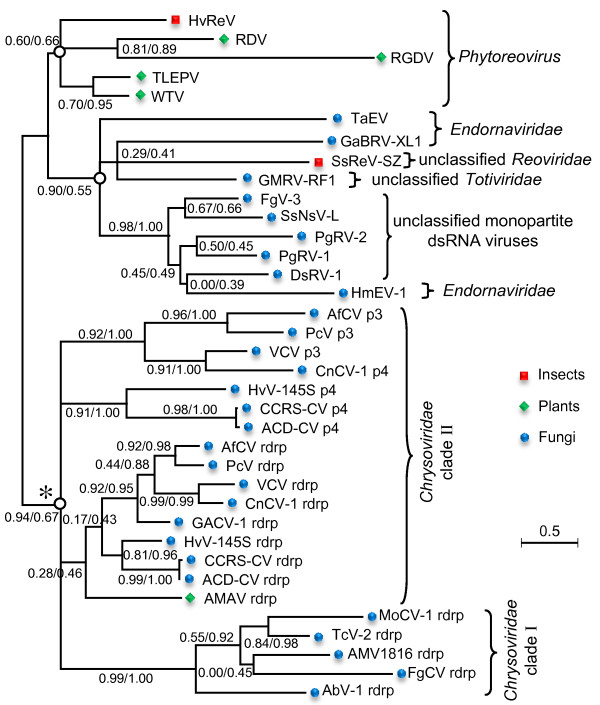
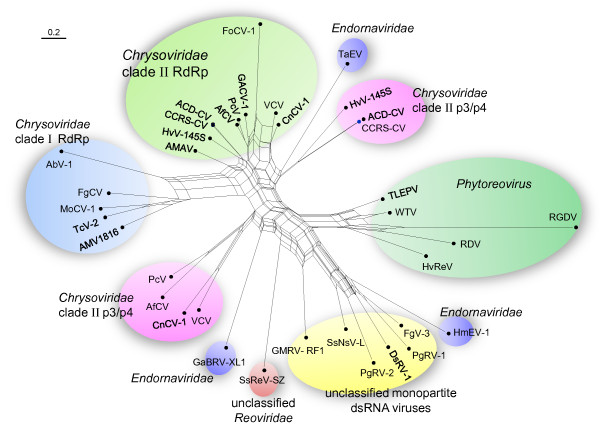
Results: In this study, we report the molecular properties of two novel dsRNA mycoviruses that were isolated from a field strain of Sclerotinia sclerotiorum, Sunf-M: one is a large monopartite virus representing a distinct evolutionary lineage of dsRNA viruses; the other is a new member of the family Partitiviridae. Comprehensive phylogenetic analysis and genome comparison revealed that there are at least ten monopartite, three bipartite, one tripartite and three quadripartite lineages in the known dsRNA mycoviruses and that the multipartite lineages have possibly evolved from different monopartite dsRNA viruses. Moreover, we found that homologs of the S7 Domain, characteristic of members of the genus phytoreovirus in family Reoviridae are widely distributed in diverse dsRNA viral lineages, including chrysoviruses, endornaviruses and some unclassified dsRNA mycoviruses. We further provided evidence that multiple HGT events may have occurred among these dsRNA viruses from different families.
Conclusions: Our study provides an insight into the phylogeny and evolution of mycovirus-related dsRNA viruses and reveals that the occurrence of HGT between different virus species and the development of multipartite genomes during evolution are important macroevolutionary mechanisms in dsRNA viruses.
Figures
References
-
- King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (Eds): Virus taxonomy: Ninth Report of the International Committee on taxonomy of viruses. New York: Elsevier; 2011.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
