

Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill
- PMID: 22717885
- PMCID: PMC3498917
- DOI: 10.1038/ismej.2012.59
Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill
Abstract
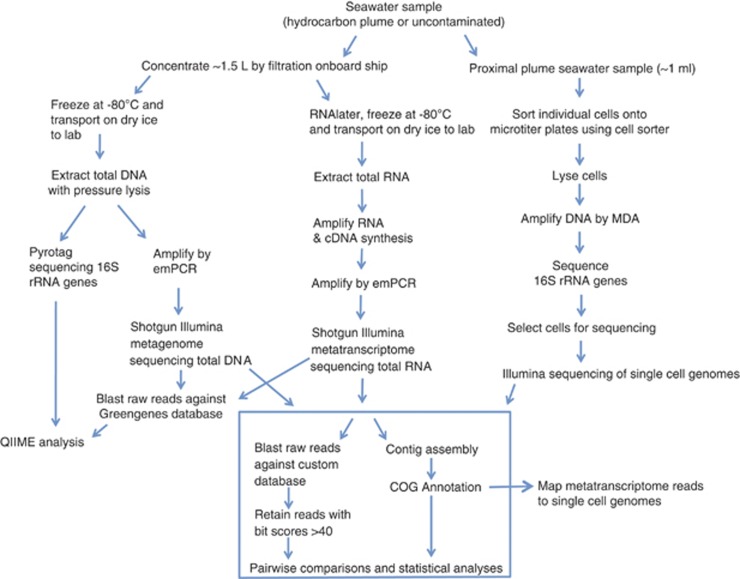
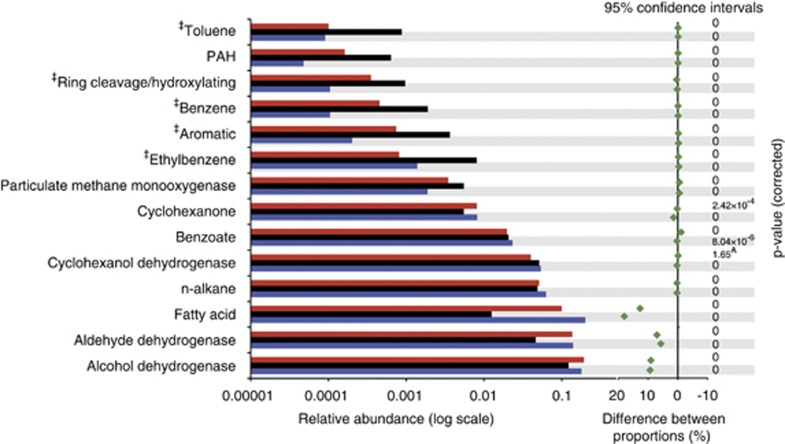
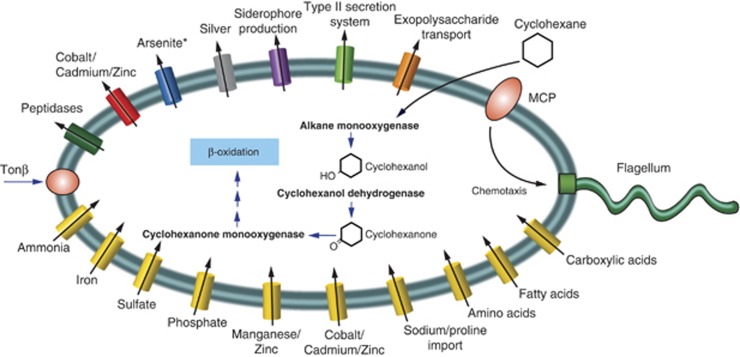
The Deepwater Horizon oil spill in the Gulf of Mexico resulted in a deep-sea hydrocarbon plume that caused a shift in the indigenous microbial community composition with unknown ecological consequences. Early in the spill history, a bloom of uncultured, thus uncharacterized, members of the Oceanospirillales was previously detected, but their role in oil disposition was unknown. Here our aim was to determine the functional role of the Oceanospirillales and other active members of the indigenous microbial community using deep sequencing of community DNA and RNA, as well as single-cell genomics. Shotgun metagenomic and metatranscriptomic sequencing revealed that genes for motility, chemotaxis and aliphatic hydrocarbon degradation were significantly enriched and expressed in the hydrocarbon plume samples compared with uncontaminated seawater collected from plume depth. In contrast, although genes coding for degradation of more recalcitrant compounds, such as benzene, toluene, ethylbenzene, total xylenes and polycyclic aromatic hydrocarbons, were identified in the metagenomes, they were expressed at low levels, or not at all based on analysis of the metatranscriptomes. Isolation and sequencing of two Oceanospirillales single cells revealed that both cells possessed genes coding for n-alkane and cycloalkane degradation. Specifically, the near-complete pathway for cyclohexane oxidation in the Oceanospirillales single cells was elucidated and supported by both metagenome and metatranscriptome data. The draft genome also included genes for chemotaxis, motility and nutrient acquisition strategies that were also identified in the metagenomes and metatranscriptomes. These data point towards a rapid response of members of the Oceanospirillales to aliphatic hydrocarbons in the deep sea.
Figures


References
-
- Camilli R, Reddy CM, Yoerger DR, Van Mooy BA, Jakuba MV, Kinsey JC, et al. Tracking hydrocarbon plume transport and biodegradation at Deepwater Horizon. Science. 2010;330:201–204. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
