

Human ZMPSTE24 disease mutations: residual proteolytic activity correlates with disease severity
- PMID: 22718200
- PMCID: PMC3428156
- DOI: 10.1093/hmg/dds233
Human ZMPSTE24 disease mutations: residual proteolytic activity correlates with disease severity
Abstract
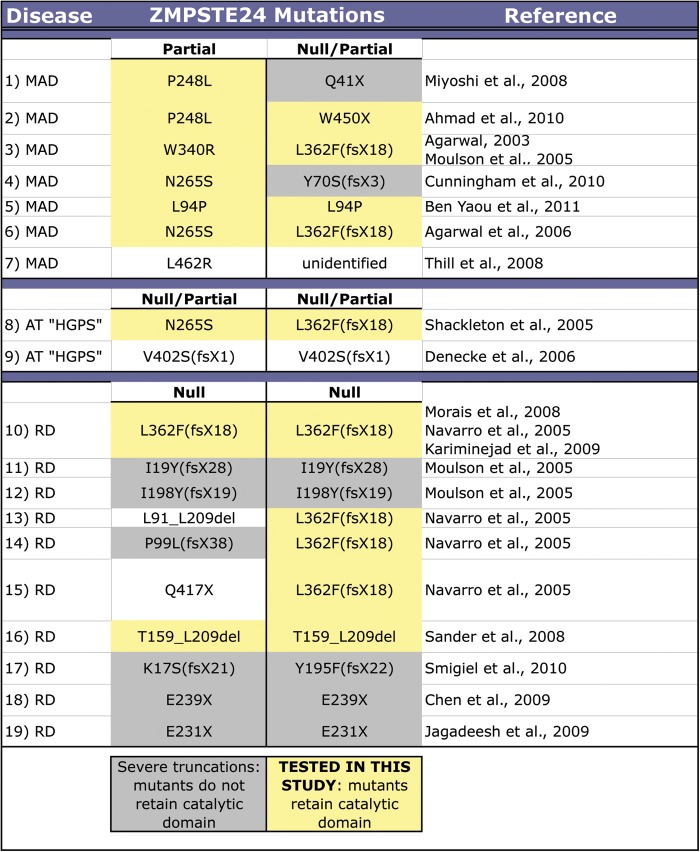
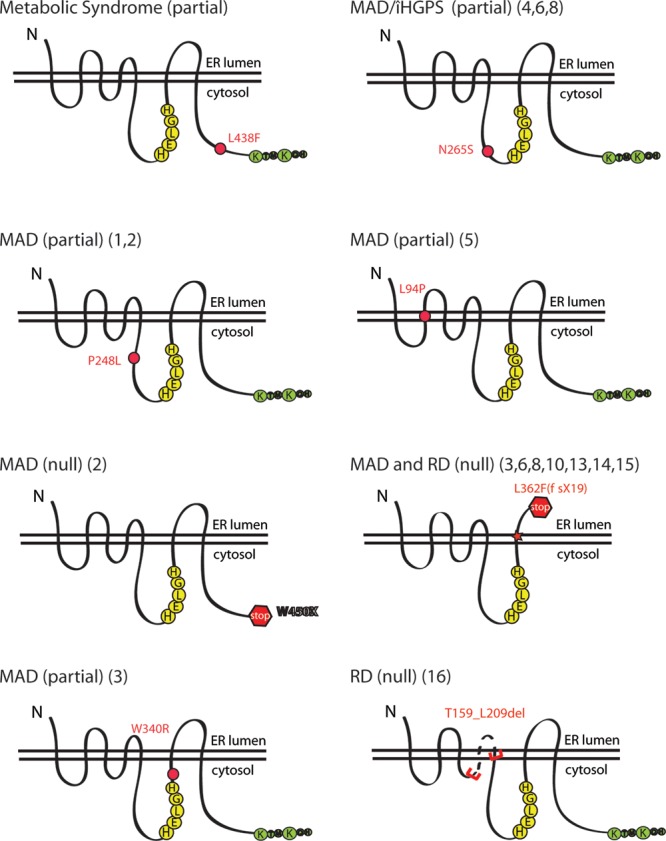
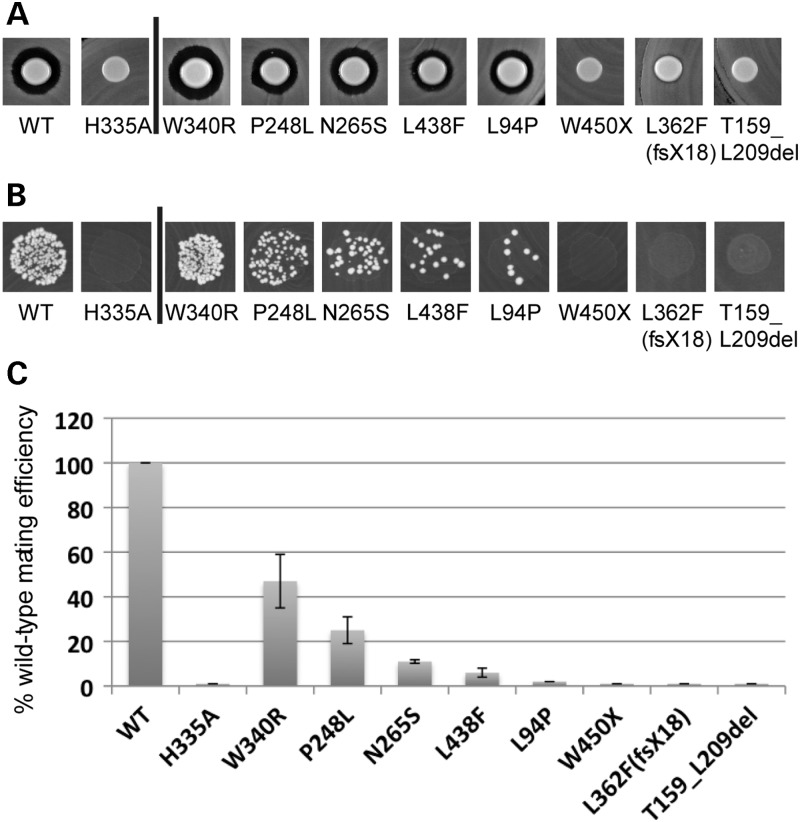
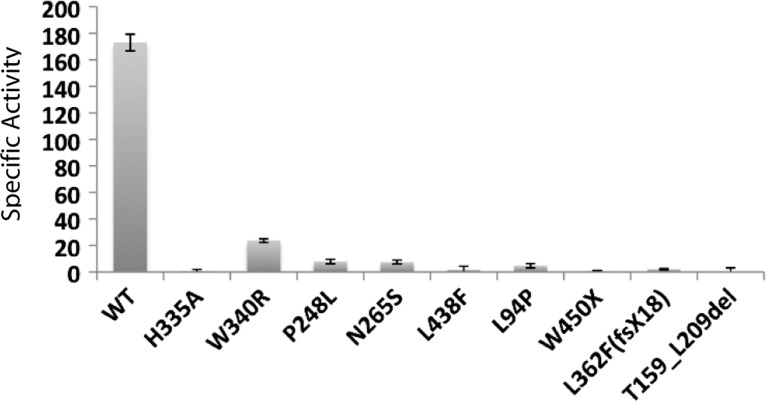
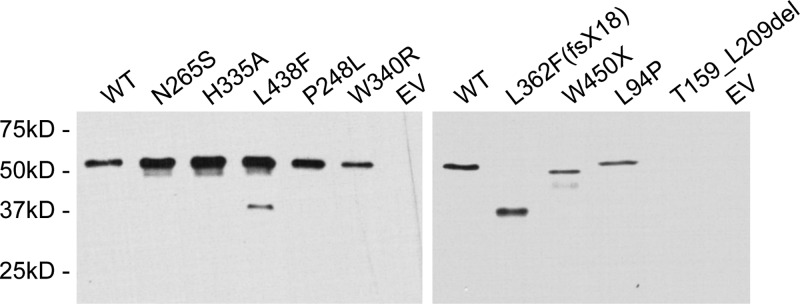
The zinc metalloprotease ZMPSTE24 plays a critical role in nuclear lamin biology by cleaving the prenylated and carboxylmethylated 15-amino acid tail from the C-terminus of prelamin A to yield mature lamin A. A defect in this proteolytic event, caused by a mutation in the lamin A gene (LMNA) that eliminates the ZMPSTE24 cleavage site, underlies the premature aging disease Hutchinson-Gilford Progeria Syndrome (HGPS). Likewise, mutations in the ZMPSTE24 gene that result in decreased enzyme function cause a spectrum of diseases that share certain features of premature aging. Twenty human ZMPSTE24 alleles have been identified that are associated with three disease categories of increasing severity: mandibuloacral dysplasia type B (MAD-B), severe progeria (atypical 'HGPS') and restrictive dermopathy (RD). To determine whether a correlation exists between decreasing ZMPSTE24 protease activity and increasing disease severity, we expressed mutant alleles of ZMPSTE24 in yeast and optimized in vivo yeast mating assays to directly compare the activity of alleles associated with each disease category. We also measured the activity of yeast crude membranes containing the ZMPSTE24 mutant proteins in vitro. We determined that, in general, the residual activity of ZMPSTE24 patient alleles correlates with disease severity. Complete loss-of-function alleles are associated with RD, whereas retention of partial, measureable activity results in MAD-B or severe progeria. Importantly, our assays can discriminate small differences in activity among the mutants, confirming that the methods presented here will be useful for characterizing any new ZMPSTE24 mutations that are discovered.
Figures

References
-
- Barrowman J., Michaelis S. ZMPSTE24, an integral membrane zinc metalloprotease with a connection to progeroid disorders. Biol. Chem. 2009;390:761–773. - PubMed
-
- Pendas A.M., Zhou Z., Cadinanos J., Freije J.M., Wang J., Hultenby K., Astudillo A., Wernerson A., Rodriguez F., Tryggvason K., et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 2002;31:94–99. - PubMed
-
- Young S.G., Fong L.G., Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria–new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J. Lipid Res. 2005;46:2531–2558. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
