

S100P-derived RAGE antagonistic peptide reduces tumor growth and metastasis
- PMID: 22718861
- PMCID: PMC3845828
- DOI: 10.1158/1078-0432.CCR-12-0221
S100P-derived RAGE antagonistic peptide reduces tumor growth and metastasis
Abstract
Purpose: The receptor for advanced glycation end products (RAGE) contributes to multiple pathologies, including diabetes, arthritis, neurodegenerative diseases, and cancer. Despite the obvious need, no RAGE inhibitors are in common clinical use. Therefore, we developed a novel small RAGE antagonist peptide (RAP) that blocks activation by multiple ligands.
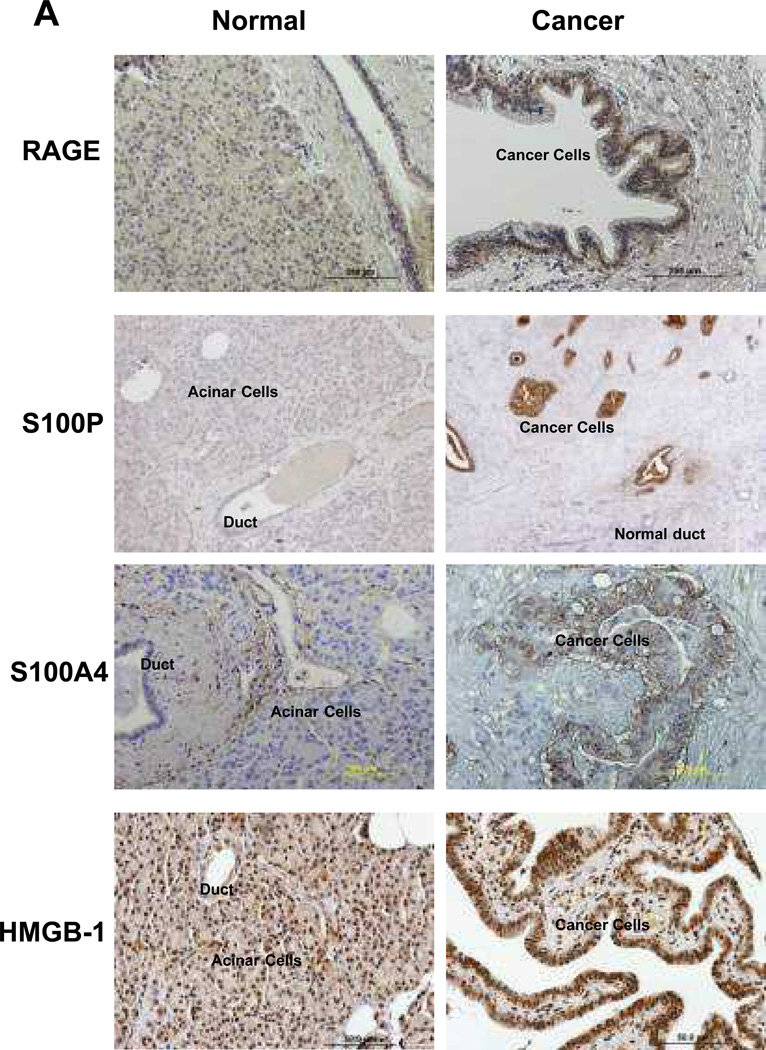
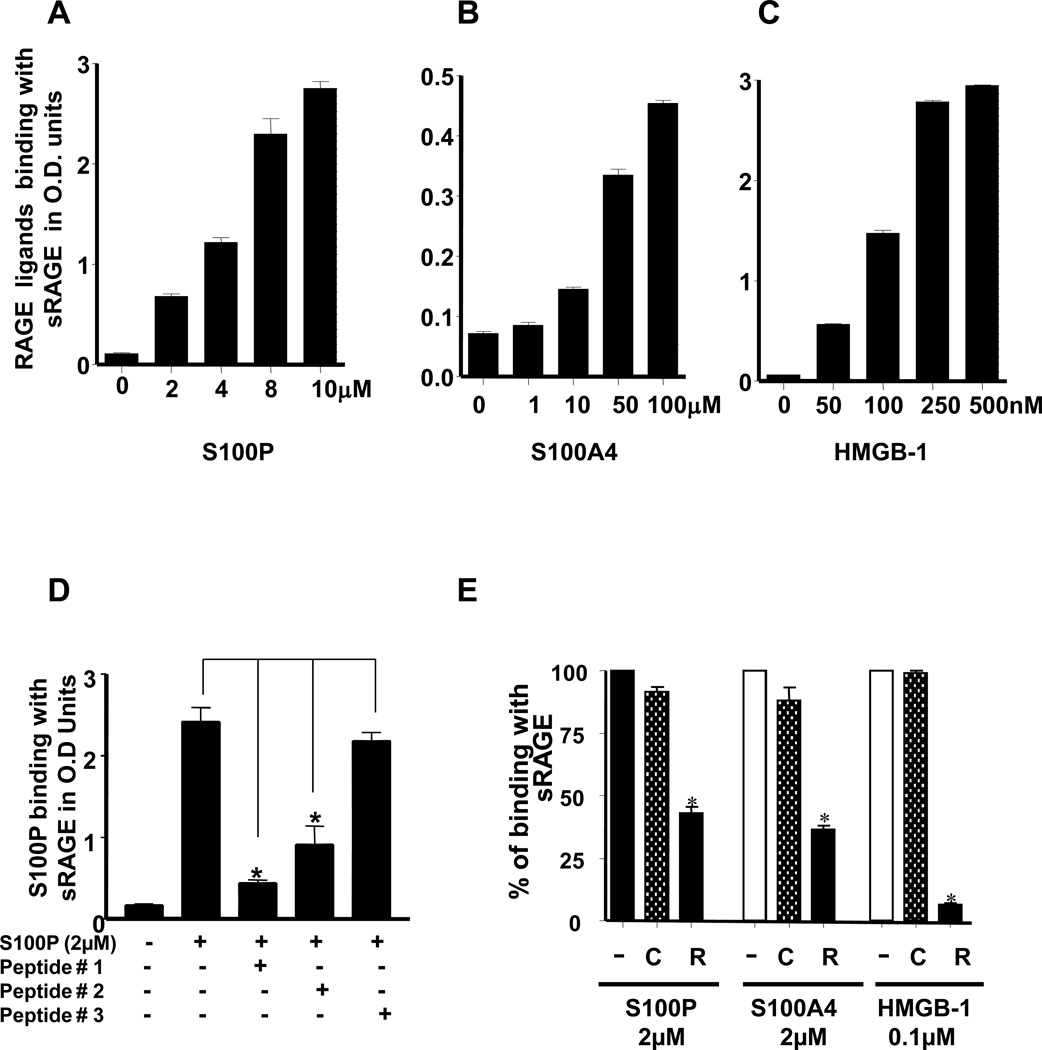
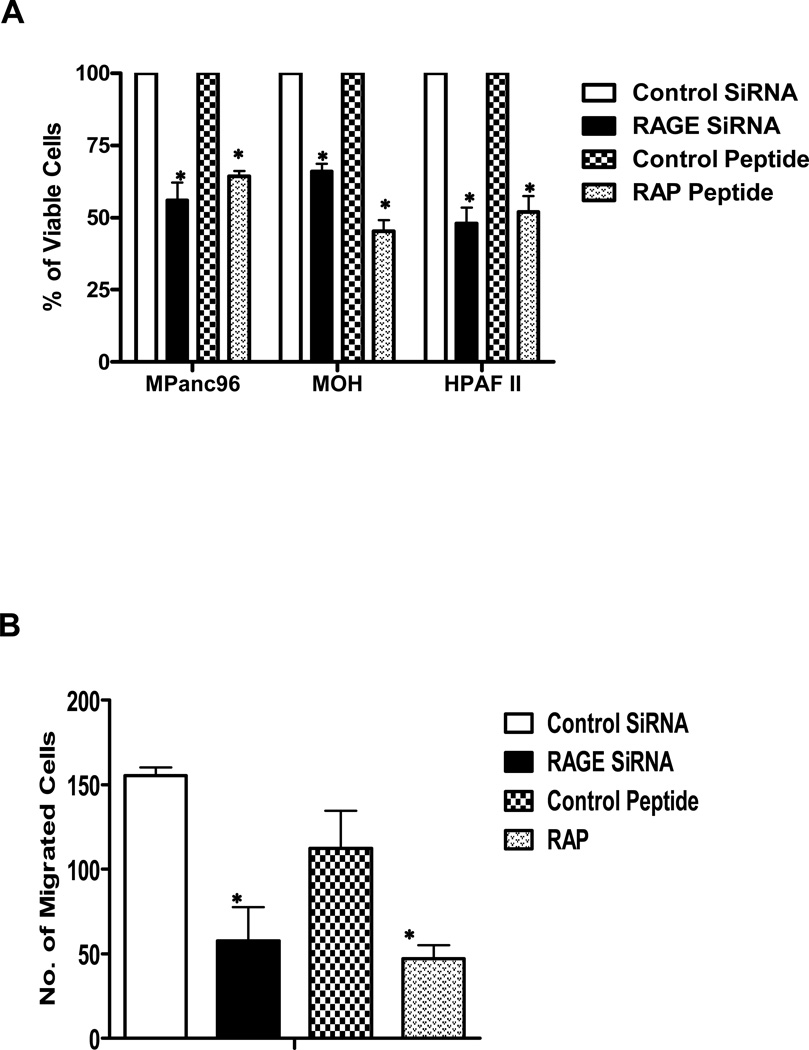
Experimental design: RAGE and its ligands were visualized by immunohistochemical analysis of human pancreatic tissues, and siRNA was used to analyze their functions. Interactions between RAGE and S100P, S100A4, and HMGB-1 were measured by ELISA. Three S100P-derived small antagonistic peptides were designed, synthesized, and tested for inhibition of RAGE binding. The effects of the peptide blockers on NFκB-luciferase reporter activity was used to assess effects on RAGE-mediated signaling. The most effective peptide was tested on glioma and pancreatic ductal adenocarcinoma (PDAC) models.
Results: Immunohistochemical analysis confirmed the expression of RAGE and its ligands S100P, S100A4, and HMGB-1 in human PDAC. siRNA silencing of RAGE or its ligands reduced the growth and migration of PDAC cells in vitro. The most effective RAP inhibited the interaction of S100P, S100A4, and HMGB-1 with RAGE at micromolar concentrations. RAP also reduced the ability of the ligands to stimulate RAGE activation of NFκB in cancer cells in vitro and in vivo. Importantly, systemic in vivo administration of RAP reduced the growth and metastasis of pancreatic tumors and also inhibited glioma tumor growth.
Conclusion: RAP shows promise as a tool for the investigation of RAGE function and as an in vivo treatment for RAGE-related disorders.
Figures






References
-
- Logsdon CD, Fuentes MK, Huang EH, Arumugam T. RAGE and RAGE ligands in cancer. Curr Mol Med. 2007;7(8):777–789. - PubMed
-
- Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. 2005;15:16R–28R. - PubMed
-
- Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005;83:876–886. - PubMed
-
- Bierhaus A, Stern DM, Nawroth PP. RAGE in inflammation: a new therapeutic target? Curr Opin Investig Drugs. 2006;7:985–991. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
