

Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans
- PMID: 22718972
- PMCID: PMC3439916
- DOI: 10.1093/nar/gks532
Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans
Abstract
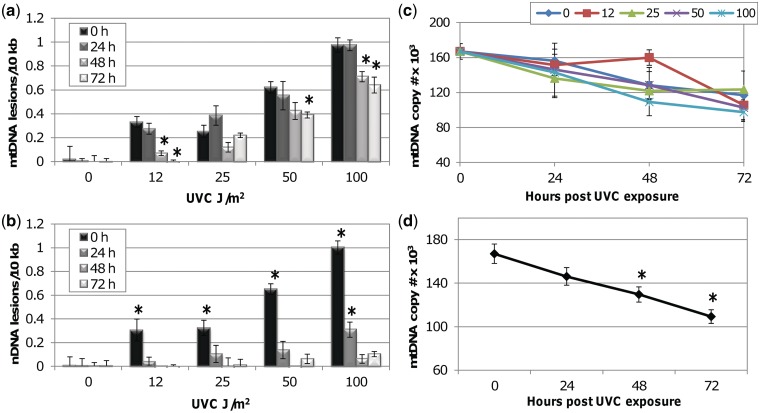
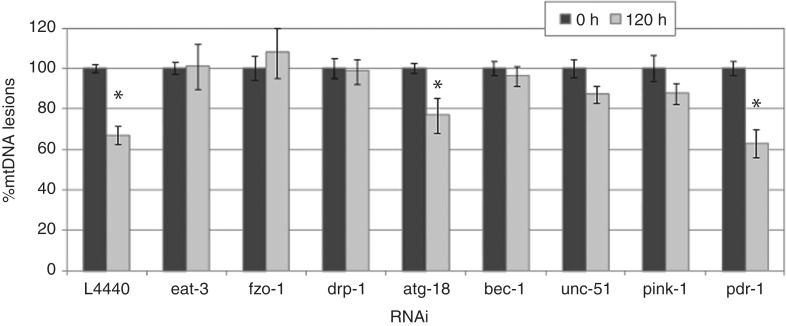
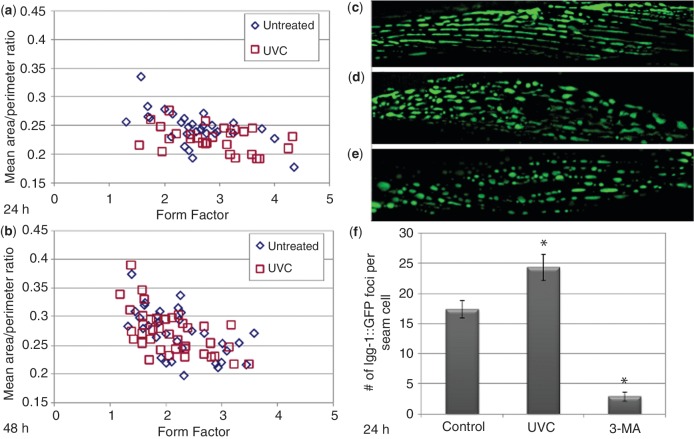
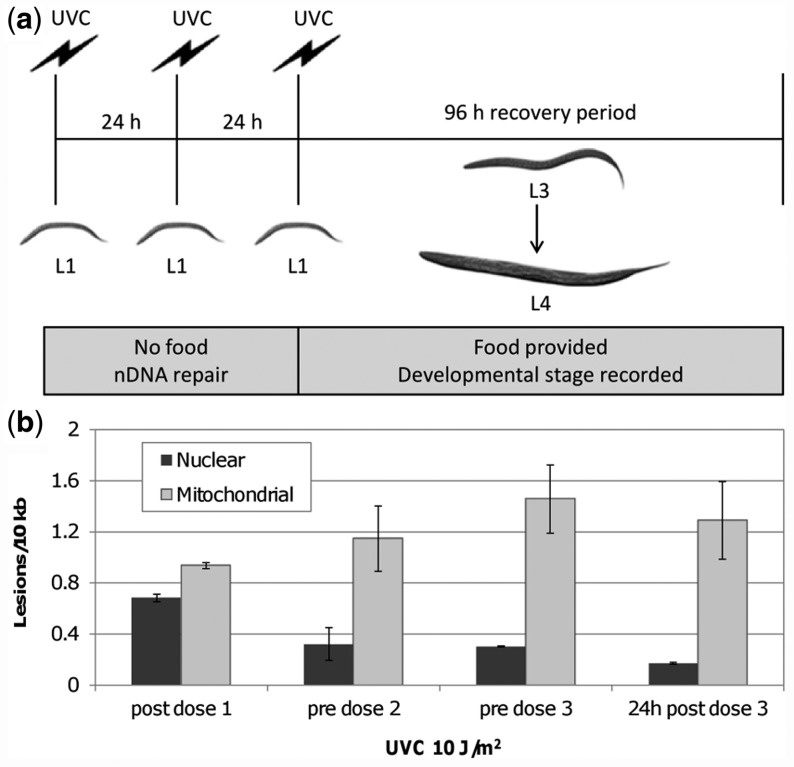
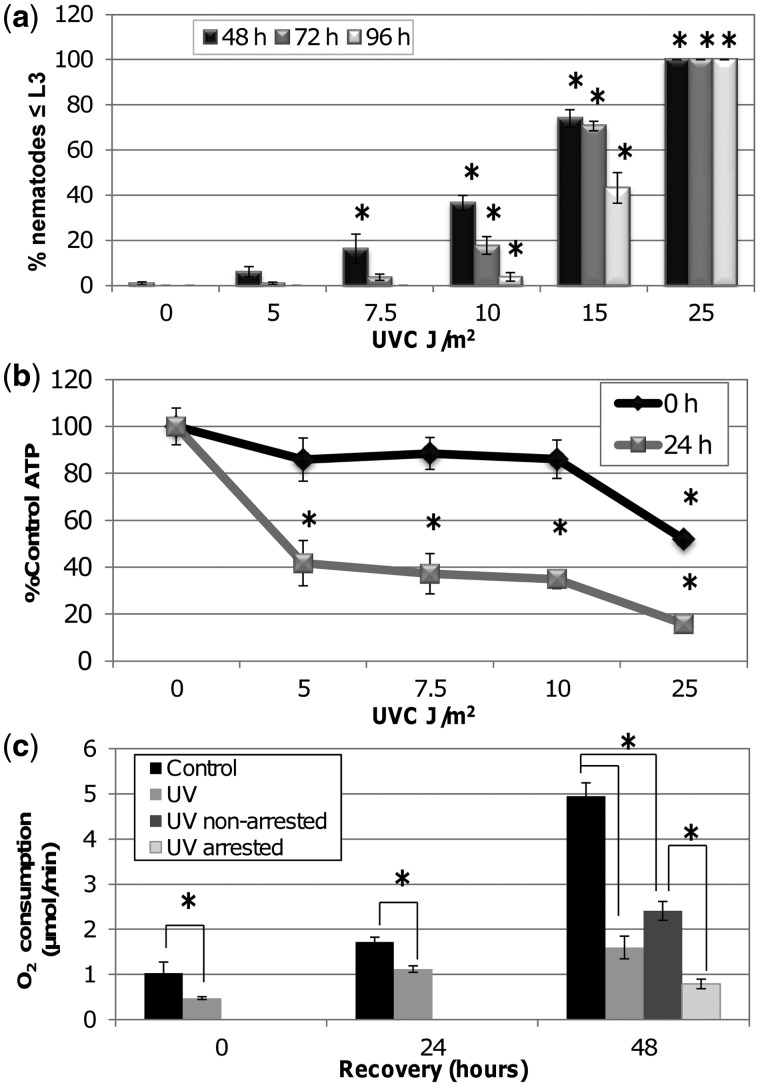
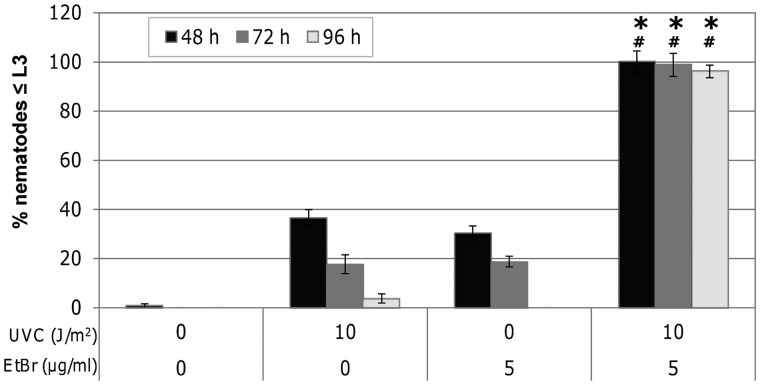
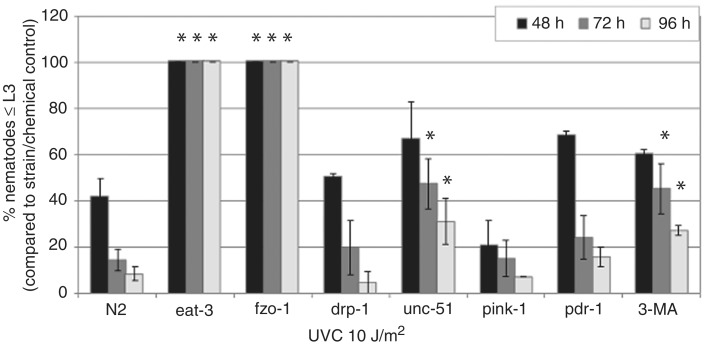
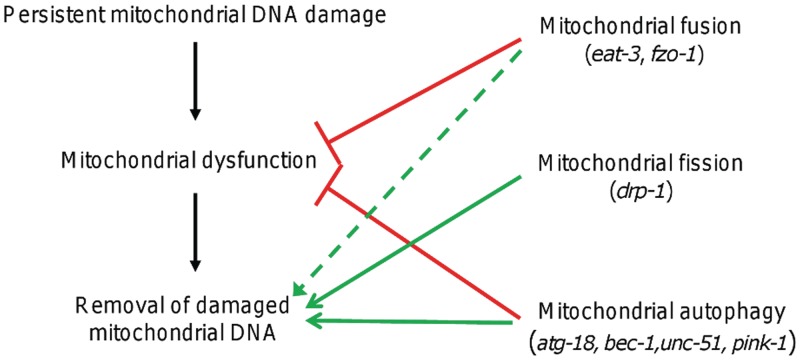
Mitochondria lack the ability to repair certain helix-distorting lesions that are induced at high levels in mitochondrial DNA (mtDNA) by important environmental genotoxins and endogenous metabolites. These lesions are irreparable and persistent in the short term, but their long-term fate is unknown. We report that removal of such mtDNA damage is detectable by 48 h in Caenorhabditis elegans, and requires mitochondrial fusion, fission and autophagy, providing genetic evidence for a novel mtDNA damage removal pathway. Furthermore, mutations in genes involved in these processes as well as pharmacological inhibition of autophagy exacerbated mtDNA damage-mediated larval arrest, illustrating the in vivo relevance of removal of persistent mtDNA damage. Mutations in genes in these pathways exist in the human population, demonstrating the potential for important gene-environment interactions affecting mitochondrial health after genotoxin exposure.
Figures
Similar articles
-
UVC-induced mitochondrial degradation via autophagy correlates with mtDNA damage removal in primary human fibroblasts.J Biochem Mol Toxicol. 2013 Jan;27(1):28-41. doi: 10.1002/jbt.21440. Epub 2012 Nov 6. J Biochem Mol Toxicol. 2013. PMID: 23132756 Free PMC article.
-
Involvement of autophagy and mitochondrial dynamics in determining the fate and effects of irreparable mitochondrial DNA damage.Autophagy. 2012 Dec;8(12):1822-23. doi: 10.4161/auto.21741. Autophagy. 2012. PMID: 22929123 Free PMC article.
-
Nonselective autophagy reduces mitochondrial content during starvation in Caenorhabditis elegans.Am J Physiol Cell Physiol. 2018 Dec 1;315(6):C781-C792. doi: 10.1152/ajpcell.00109.2018. Epub 2018 Aug 22. Am J Physiol Cell Physiol. 2018. PMID: 30133321 Free PMC article.
-
Mitochondrial fusion, fission, and mitochondrial toxicity.Toxicology. 2017 Nov 1;391:42-53. doi: 10.1016/j.tox.2017.07.019. Epub 2017 Aug 5. Toxicology. 2017. PMID: 28789970 Free PMC article. Review.
-
Mitochondrial DNA damage and its consequences for mitochondrial gene expression.Biochim Biophys Acta. 2012 Sep-Oct;1819(9-10):979-91. doi: 10.1016/j.bbagrm.2012.06.002. Epub 2012 Jun 19. Biochim Biophys Acta. 2012. PMID: 22728831 Free PMC article. Review.
Cited by
-
In vivo repair of alkylating and oxidative DNA damage in the mitochondrial and nuclear genomes of wild-type and glycosylase-deficient Caenorhabditis elegans.DNA Repair (Amst). 2012 Nov 1;11(11):857-63. doi: 10.1016/j.dnarep.2012.08.002. Epub 2012 Sep 5. DNA Repair (Amst). 2012. PMID: 22959841 Free PMC article.
-
Deleterious mitochondrial DNA point mutations are overrepresented in Drosophila expressing a proofreading-defective DNA polymerase γ.PLoS Genet. 2018 Nov 19;14(11):e1007805. doi: 10.1371/journal.pgen.1007805. eCollection 2018 Nov. PLoS Genet. 2018. PMID: 30452458 Free PMC article.
-
Characterization of HBV integration patterns and timing in liver cancer and HBV-infected livers.Oncotarget. 2018 May 18;9(38):25075-25088. doi: 10.18632/oncotarget.25308. eCollection 2018 May 18. Oncotarget. 2018. PMID: 29861854 Free PMC article.
-
Effects of 5'-fluoro-2-deoxyuridine on mitochondrial biology in Caenorhabditis elegans.Exp Gerontol. 2014 Aug;56:69-76. doi: 10.1016/j.exger.2014.03.021. Epub 2014 Apr 3. Exp Gerontol. 2014. PMID: 24704715 Free PMC article.
-
Mitochondrial Toxicity.Toxicol Sci. 2018 Mar 1;162(1):15-23. doi: 10.1093/toxsci/kfy008. Toxicol Sci. 2018. PMID: 29340618 Free PMC article. Review.
References
-
- Greaves LC, Reeve AK, Taylor RW, Turnbull DM. Mitochondrial DNA and disease. J. Pathol. 2012;226:274–286. - PubMed
-
- Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr. Opin. Cell Biol. 2003;15:706–716. - PubMed
-
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. - PubMed
-
- Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. - PubMed
-
- Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–4662. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
