

i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules
- PMID: 22718975
- PMCID: PMC3424583
- DOI: 10.1093/nar/gks543
i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules
Abstract
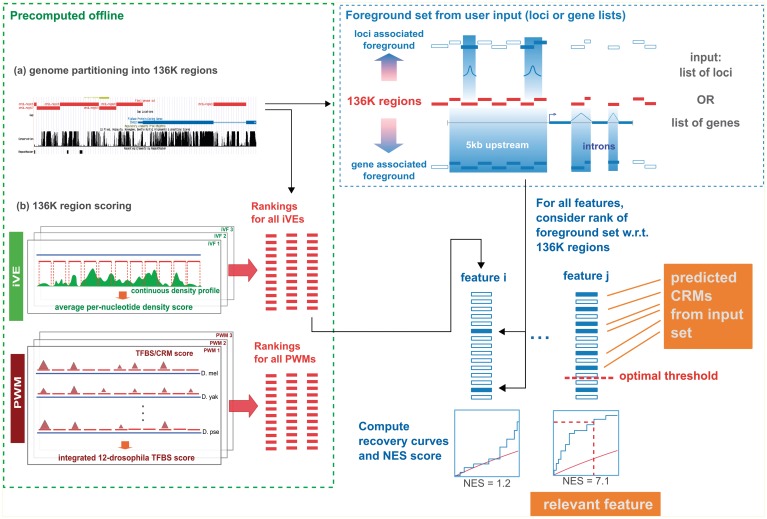
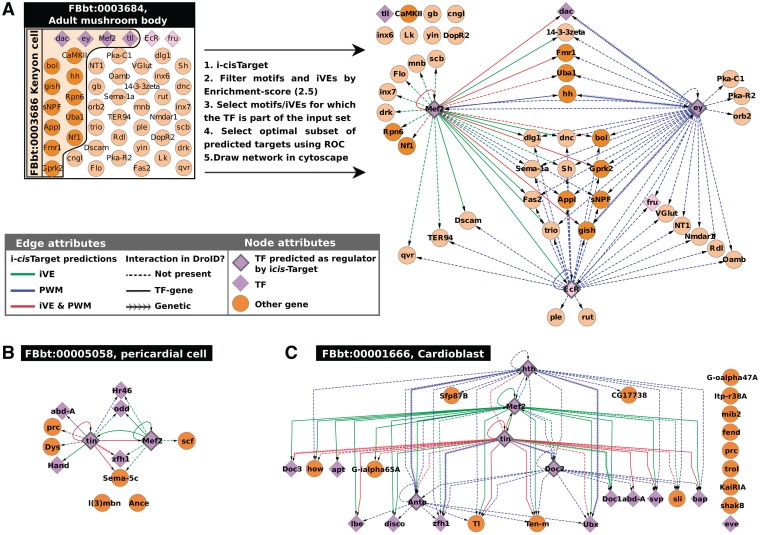
The field of regulatory genomics today is characterized by the generation of high-throughput data sets that capture genome-wide transcription factor (TF) binding, histone modifications, or DNAseI hypersensitive regions across many cell types and conditions. In this context, a critical question is how to make optimal use of these publicly available datasets when studying transcriptional regulation. Here, we address this question in Drosophila melanogaster for which a large number of high-throughput regulatory datasets are available. We developed i-cisTarget (where the 'i' stands for integrative), for the first time enabling the discovery of different types of enriched 'regulatory features' in a set of co-regulated sequences in one analysis, being either TF motifs or 'in vivo' chromatin features, or combinations thereof. We have validated our approach on 15 co-expressed gene sets, 21 ChIP data sets, 628 curated gene sets and multiple individual case studies, and show that meaningful regulatory features can be confidently discovered; that bona fide enhancers can be identified, both by in vivo events and by TF motifs; and that combinations of in vivo events and TF motifs further increase the performance of enhancer prediction.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
