

Metabolic reconstruction for metagenomic data and its application to the human microbiome
- PMID: 22719234
- PMCID: PMC3374609
- DOI: 10.1371/journal.pcbi.1002358
Metabolic reconstruction for metagenomic data and its application to the human microbiome
Abstract
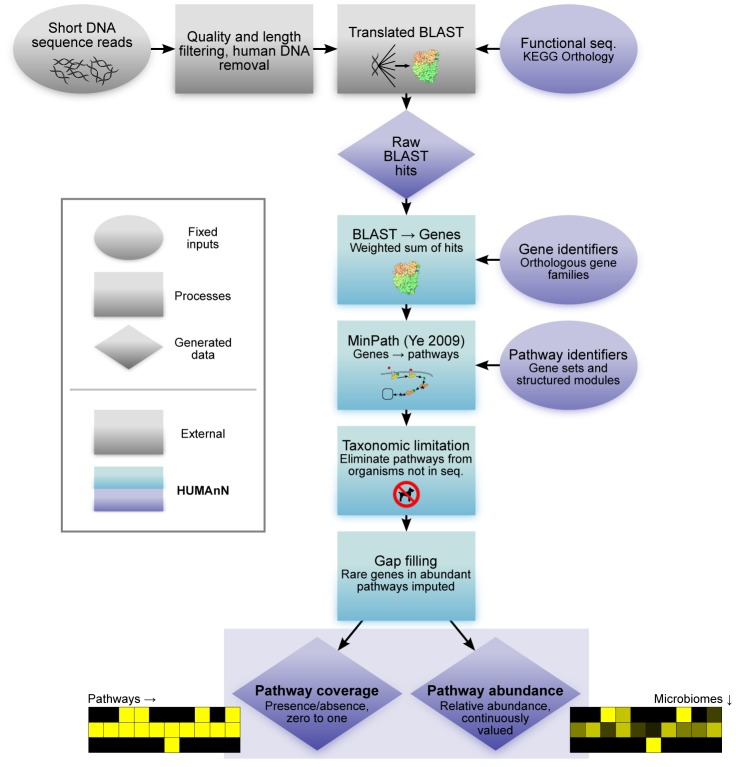
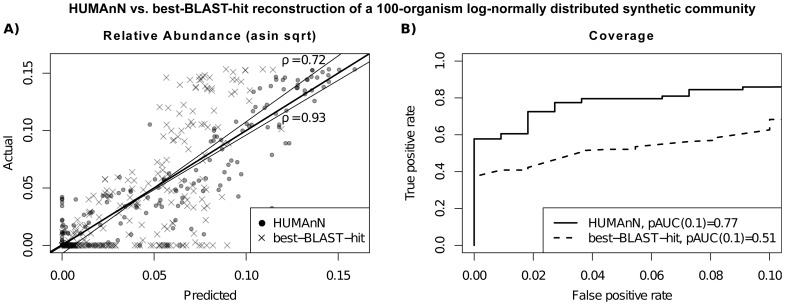
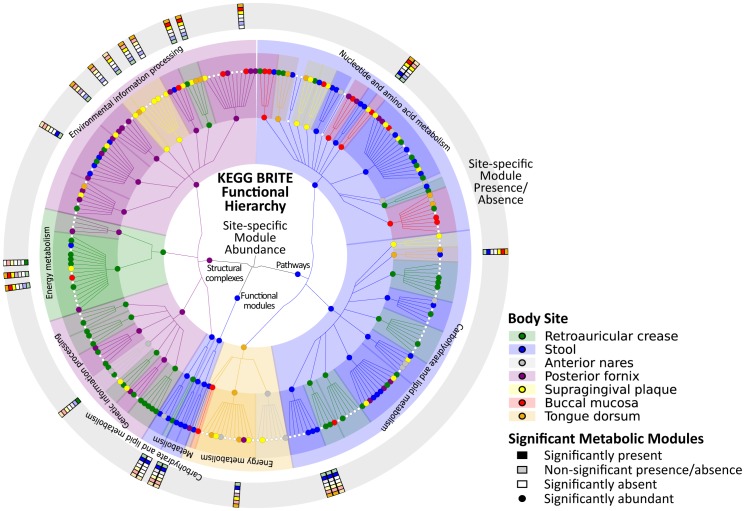
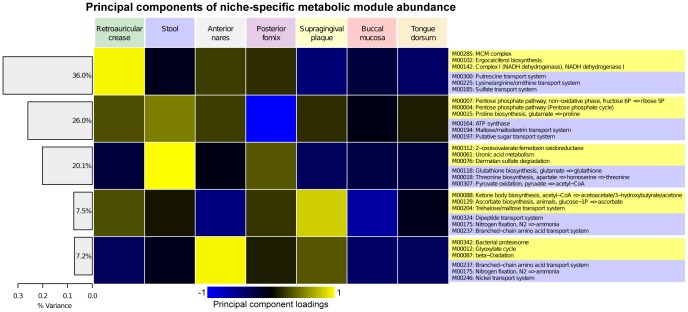
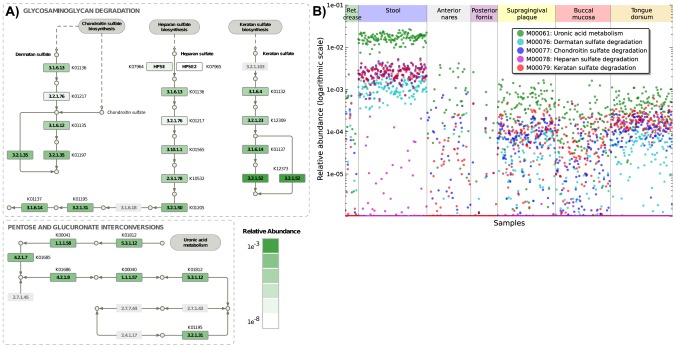
Microbial communities carry out the majority of the biochemical activity on the planet, and they play integral roles in processes including metabolism and immune homeostasis in the human microbiome. Shotgun sequencing of such communities' metagenomes provides information complementary to organismal abundances from taxonomic markers, but the resulting data typically comprise short reads from hundreds of different organisms and are at best challenging to assemble comparably to single-organism genomes. Here, we describe an alternative approach to infer the functional and metabolic potential of a microbial community metagenome. We determined the gene families and pathways present or absent within a community, as well as their relative abundances, directly from short sequence reads. We validated this methodology using a collection of synthetic metagenomes, recovering the presence and abundance both of large pathways and of small functional modules with high accuracy. We subsequently applied this method, HUMAnN, to the microbial communities of 649 metagenomes drawn from seven primary body sites on 102 individuals as part of the Human Microbiome Project (HMP). This provided a means to compare functional diversity and organismal ecology in the human microbiome, and we determined a core of 24 ubiquitously present modules. Core pathways were often implemented by different enzyme families within different body sites, and 168 functional modules and 196 metabolic pathways varied in metagenomic abundance specifically to one or more niches within the microbiome. These included glycosaminoglycan degradation in the gut, as well as phosphate and amino acid transport linked to host phenotype (vaginal pH) in the posterior fornix. An implementation of our methodology is available at http://huttenhower.sph.harvard.edu/humann. This provides a means to accurately and efficiently characterize microbial metabolic pathways and functional modules directly from high-throughput sequencing reads, enabling the determination of community roles in the HMP cohort and in future metagenomic studies.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- The Human Microbiome Consortium. Structure, Function and Diversity of the Human Microbiome in an Adult Reference Population. Nature. E-pub ahead of print. 2012. doi: 10.1038/nature11234. - DOI
-
- Stecher B, Hardt WD. The role of microbiota in infectious disease. Trends Microbiol. 2008;16:114. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HG005969/HG/NHGRI NIH HHS/United States
- R21 DE017106/DE/NIDCR NIH HHS/United States
- CA139193/CA/NCI NIH HHS/United States
- U54HG004968/HG/NHGRI NIH HHS/United States
- 5R01HG005975/HG/NHGRI NIH HHS/United States
- U54HG004969/HG/NHGRI NIH HHS/United States
- T32 AI007528/AI/NIAID NIH HHS/United States
- R01 HG005975/HG/NHGRI NIH HHS/United States
- U54 HG004968/HG/NHGRI NIH HHS/United States
- R21 CA139193/CA/NCI NIH HHS/United States
- 1R01HG005969/HG/NHGRI NIH HHS/United States
- DE017106/DE/NIDCR NIH HHS/United States
- U54 HG004969/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
