

Current status of treatment of spinal and bulbar muscular atrophy
- PMID: 22720173
- PMCID: PMC3376774
- DOI: 10.1155/2012/369284
Current status of treatment of spinal and bulbar muscular atrophy
Abstract
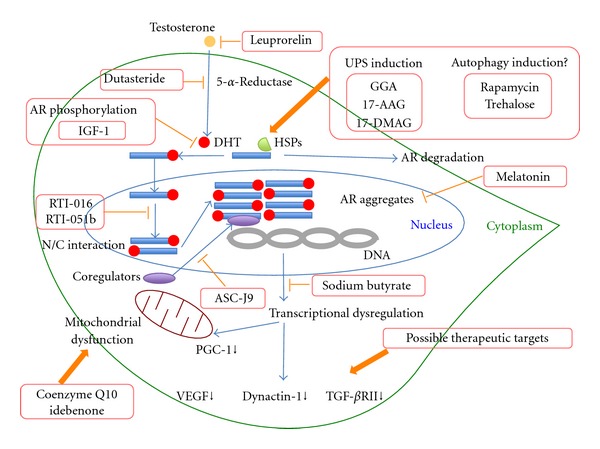
Spinal and bulbar muscular atrophy (SBMA) is the first member identified among polyglutamine diseases characterized by slowly progressive muscle weakness and atrophy of the bulbar, facial, and limb muscles pathologically associated with motor neuron loss in the spinal cord and brainstem. Androgen receptor (AR), a disease-causing protein of SBMA, is a well-characterized ligand-activated transcription factor, and androgen binding induces nuclear translocation, conformational change and recruitment of coregulators for transactivation of AR target genes. Some therapeutic strategies for SBMA are based on these native functions of AR. Since ligand-induced nuclear translocation of mutant AR has been shown to be a critical step in motor neuron degeneration in SBMA, androgen deprivation therapies using leuprorelin and dutasteride have been developed and translated into clinical trials. Although the results of these trials are inconclusive, renewed clinical trials with more sophisticated design might prove the effectiveness of hormonal intervention in the near future. Furthermore, based on the normal function of AR, therapies targeted for conformational changes of AR including amino-terminal (N) and carboxy-terminal (C) (N/C) interaction and transcriptional coregulators might be promising. Other treatments targeted for mitochondrial function, ubiquitin-proteasome system (UPS), and autophagy could be applicable for all types of polyglutamine diseases.
Figures
References
-
- Kawahara H. A family of progressive bulbar palsy. Aichi Medical Journal. 1897;16:3–4.
-
- Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968;18(7):671–680. - PubMed
-
- Parodi S, Pennuto M. Neurotoxic effects of androgens in spinal and bulbar muscular atrophy. Frontiers in Neuroendocrinology. 2011;32(4):416–425. - PubMed
-
- Katsuno M, Adachi H, Waza M, et al. Pathogenesis, animal models and therapeutics in Spinal and bulbar muscular atrophy (SBMA) Experimental Neurology. 2006;200(1):8–18. - PubMed
-
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352(6330):77–79. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
