

Axonal degeneration in Alzheimer's disease: when signaling abnormalities meet the axonal transport system
- PMID: 22721767
- PMCID: PMC3465504
- DOI: 10.1016/j.expneurol.2012.06.003
Axonal degeneration in Alzheimer's disease: when signaling abnormalities meet the axonal transport system
Abstract
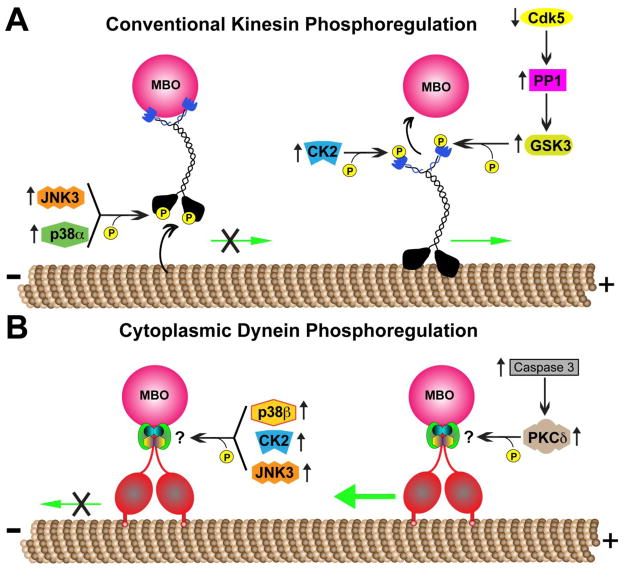
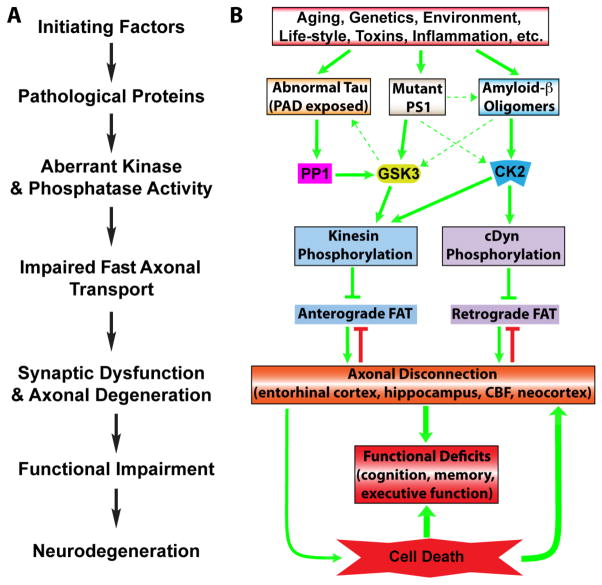
Alzheimer's disease (AD) is characterized by progressive, age-dependent degeneration of neurons in the central nervous system. A large body of evidence indicates that neurons affected in AD follow a dying-back pattern of degeneration, where abnormalities in synaptic function and axonal connectivity long precede somatic cell death. Mechanisms underlying dying-back degeneration of neurons in AD remain elusive but several have been proposed, including deficits in fast axonal transport (FAT). Accordingly, genetic evidence linked alterations in FAT to dying-back degeneration of neurons, and FAT defects have been widely documented in various AD models. In light of these findings, we discuss experimental evidence linking several AD-related pathogenic polypeptides to aberrant activation of signaling pathways involved in the phosphoregulation of microtubule-based motor proteins. While each pathway appears to affect FAT in a unique manner, in the context of AD, many of these pathways might work synergistically to compromise the delivery of molecular components critical for the maintenance and function of synapses and axons. Therapeutic approaches aimed at preventing FAT deficits by normalizing the activity of specific protein kinases may help prevent degeneration of vulnerable neurons in AD.
Keywords: Alzheimer's disease; Amyloid; ApoE; Axonal transport; CK2; Dynein; GSK3; Kinase; Kinesin; Phosphatase; Presenilin; Signaling; Synapse; Tau.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Beffert U, Morfini G, Bock HH, Reyna H, Brady ST, Herz J. Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. J Biol Chem. 2002;277:49958–49964. - PubMed
Publication types
MeSH terms
Grants and funding
- R56 NS023868/NS/NINDS NIH HHS/United States
- RC1 AG036208/AG/NIA NIH HHS/United States
- P01 AG009466/AG/NIA NIH HHS/United States
- AG033570/AG/NIA NIH HHS/United States
- AG09466/AG/NIA NIH HHS/United States
- T32 AG020506-07/AG/NIA NIH HHS/United States
- R01 AG033570/AG/NIA NIH HHS/United States
- RF1 AG033570/AG/NIA NIH HHS/United States
- T32 AG020506/AG/NIA NIH HHS/United States
- 1RC1AG036208/AG/NIA NIH HHS/United States
- NS23868/NS/NINDS NIH HHS/United States
- NS066942A/NS/NINDS NIH HHS/United States
- R01 NS023868/NS/NINDS NIH HHS/United States
- R01 NS066942/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
