

Novel 9q34.11 gene deletions encompassing combinations of four Mendelian disease genes: STXBP1, SPTAN1, ENG, and TOR1A
- PMID: 22722545
- PMCID: PMC3713627
- DOI: 10.1038/gim.2012.65
Novel 9q34.11 gene deletions encompassing combinations of four Mendelian disease genes: STXBP1, SPTAN1, ENG, and TOR1A
Abstract
Purpose: A number of genes in the 9q34.11 region may be haploinsufficient. However, studies analyzing genotype-phenotype correlations of deletions encompassing multiple dosage-sensitive genes in the region are lacking.
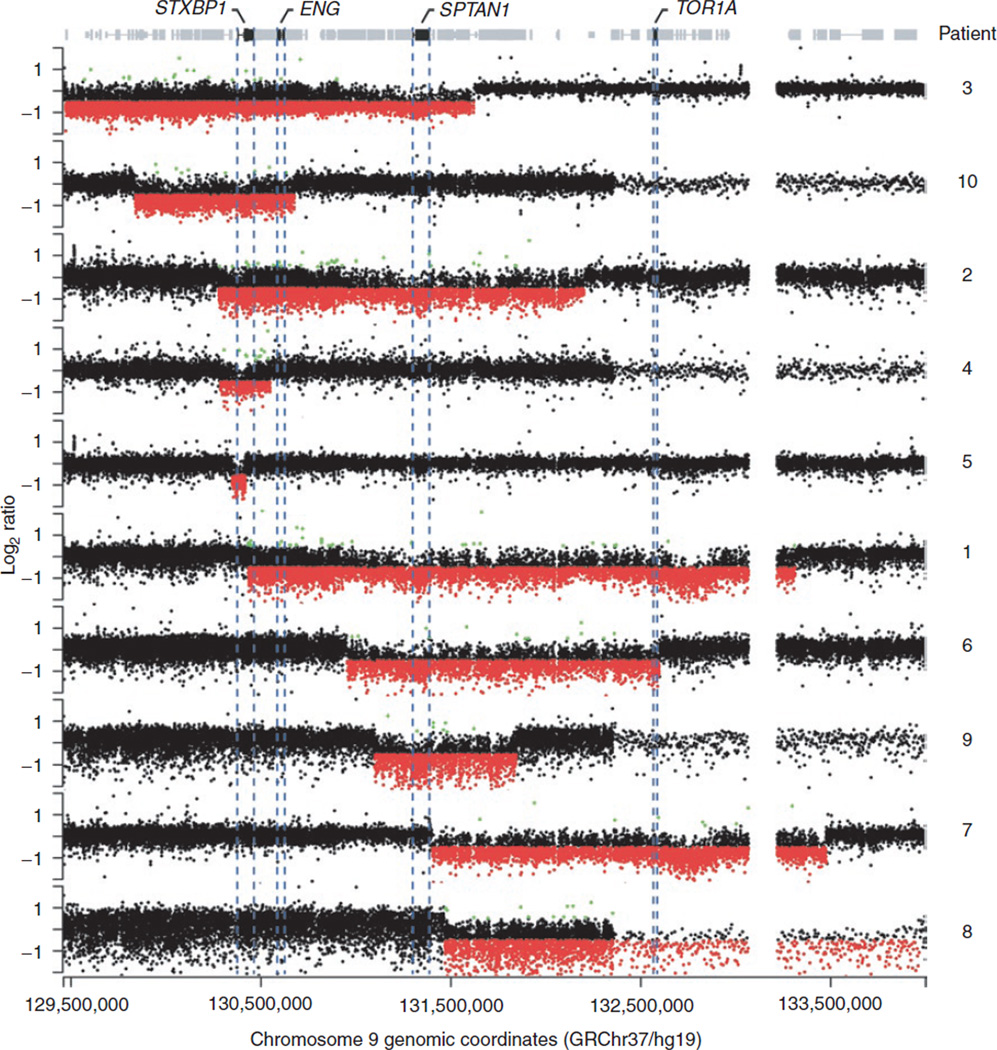
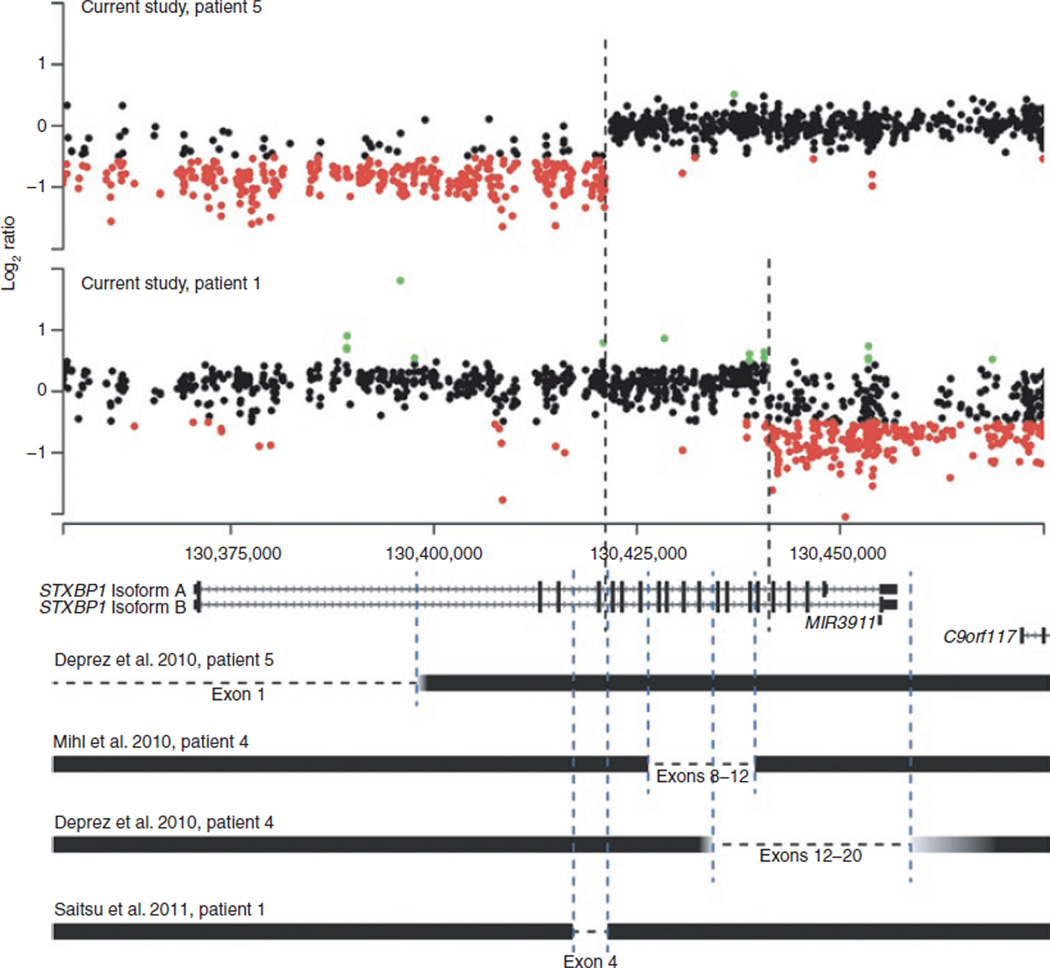
Methods: We mapped breakpoints of 10 patients with 9q34.11 deletions using high-resolution 9q34-specific array comparative genomic hybridization (CGH) to determine deletion size and gene content.
Results: The 9q34.11 deletions range in size from 67 kb to 2.8 Mb. Six patients exhibit intellectual disability and share a common deleted region including STXBP1; four manifest variable epilepsy. In five subjects, deletions include SPTAN1, previously associated with early infantile epileptic encephalopathy, infantile spasms, intellectual disability, and hypomyelination. In four patients, the deletion includes endoglin (ENG), causative of hereditary hemorrhagic telangiectasia. Finally, in four patients, deletions involve TOR1A, of which molecular defects lead to early-onset primary dystonia. Ninety-four other RefSeq genes also map to the genomic intervals investigated.
Conclusion: STXBP1 haploinsufficiency results in progressive encephalopathy characterized by intellectual disability and may be accompanied by epilepsy, movement disorders, and autism. We propose that 9q34.11 genomic deletions involving ENG, TOR1A, STXBP1, and SPTAN1 are responsible for multisystemic vascular dysplasia, early-onset primary dystonia, epilepsy, and intellectual disability, therefore revealing cis-genetic effects leading to complex phenotypes.
Figures
References
-
- Ohtahara S, Ohtsuka Y, Yamatogi Y, Oka E. The early-infantile epileptic encephalopathy with suppression-burst: developmental aspects. Brain Dev. 1987;9:371–376. - PubMed
-
- Zupanc ML. Clinical evaluation and diagnosis of severe epilepsy syndromes of early childhood. J Child Neurol. 2009;24(8 Suppl):6S–14S. - PubMed
-
- Ohtahara S, Yamatogi Y. Epileptic encephalopathies in early infancy with suppression-burst. J Clin Neurophysiol. 2003;20:398–407. - PubMed
-
- Strømme P, Mangelsdorf ME, Shaw MA, et al. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet. 2002;30:441–445. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
