

The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers
- PMID: 22722843
- PMCID: PMC3436069
- DOI: 10.1038/nature11219
The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers
Abstract
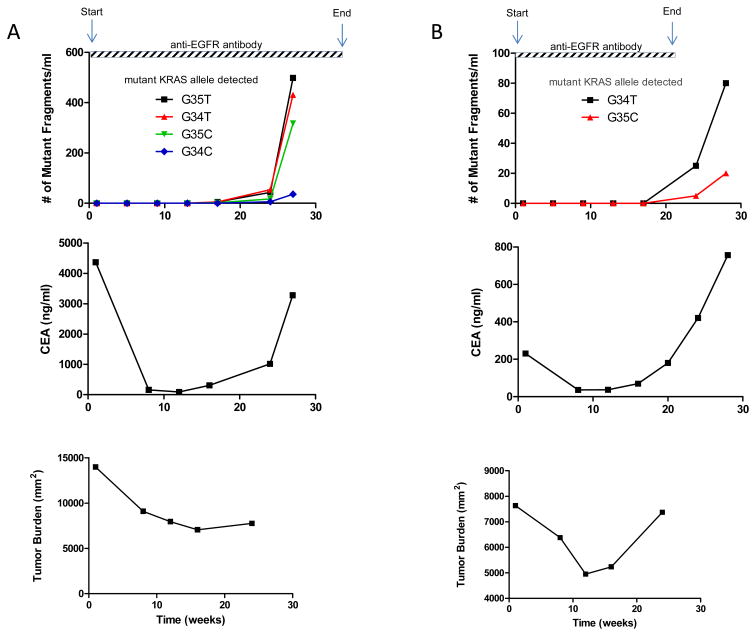
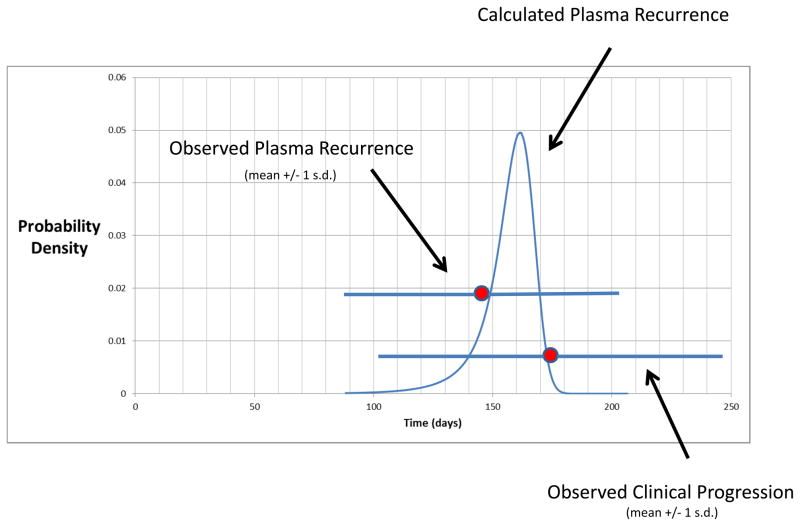
Colorectal tumours that are wild type for KRAS are often sensitive to EGFR blockade, but almost always develop resistance within several months of initiating therapy. The mechanisms underlying this acquired resistance to anti-EGFR antibodies are largely unknown. This situation is in marked contrast to that of small-molecule targeted agents, such as inhibitors of ABL, EGFR, BRAF and MEK, in which mutations in the genes encoding the protein targets render the tumours resistant to the effects of the drugs. The simplest hypothesis to account for the development of resistance to EGFR blockade is that rare cells with KRAS mutations pre-exist at low levels in tumours with ostensibly wild-type KRAS genes. Although this hypothesis would seem readily testable, there is no evidence in pre-clinical models to support it, nor is there data from patients. To test this hypothesis, we determined whether mutant KRAS DNA could be detected in the circulation of 28 patients receiving monotherapy with panitumumab, a therapeutic anti-EGFR antibody. We found that 9 out of 24 (38%) patients whose tumours were initially KRAS wild type developed detectable mutations in KRAS in their sera, three of which developed multiple different KRAS mutations. The appearance of these mutations was very consistent, generally occurring between 5 and 6 months following treatment. Mathematical modelling indicated that the mutations were present in expanded subclones before the initiation of panitumumab treatment. These results suggest that the emergence of KRAS mutations is a mediator of acquired resistance to EGFR blockade and that these mutations can be detected in a non-invasive manner. They explain why solid tumours develop resistance to targeted therapies in a highly reproducible fashion.
Figures
Comment in
-
Gastrointestinal cancer: a step closer to combating acquired resistance in CRC.Nat Rev Clin Oncol. 2012 Jun 26;9(8):428. doi: 10.1038/nrclinonc.2012.114. Nat Rev Clin Oncol. 2012. PMID: 22733232 No abstract available.
-
Cancer: Pinprick diagnostics.Nature. 2012 Jun 27;486(7404):482-3. doi: 10.1038/486482a. Nature. 2012. PMID: 22739312 No abstract available.
-
Colorectal cancer: A step closer to combating acquired resistance in CRC.Nat Rev Gastroenterol Hepatol. 2012 Aug;9(8):427. doi: 10.1038/nrgastro.2012.128. Epub 2012 Jul 3. Nat Rev Gastroenterol Hepatol. 2012. PMID: 22751457 No abstract available.
References
-
- Amado RG, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. - PubMed
-
- Karapetis CS, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. - PubMed
-
- Engelman JA, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 GM058008/GM/NIGMS NIH HHS/United States
- P50 CA062924/CA/NCI NIH HHS/United States
- R01GM078986/GM/NIGMS NIH HHS/United States
- CA62924/CA/NCI NIH HHS/United States
- N01 CN043309/CA/NCI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- CA43460/CA/NCI NIH HHS/United States
- N01-CN-43309/CN/NCI NIH HHS/United States
- R37 CA057345/CA/NCI NIH HHS/United States
- R01 CA129825/CA/NCI NIH HHS/United States
- R01 CA057345/CA/NCI NIH HHS/United States
- CA129825/CA/NCI NIH HHS/United States
- CA57345/CA/NCI NIH HHS/United States
- R37 CA043460/CA/NCI NIH HHS/United States
- P50 CA095103/CA/NCI NIH HHS/United States
- CA095103/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
