

Diversity and Distribution of Marine Synechococcus: Multiple Gene Phylogenies for Consensus Classification and Development of qPCR Assays for Sensitive Measurement of Clades in the Ocean
- PMID: 22723796
- PMCID: PMC3377940
- DOI: 10.3389/fmicb.2012.00213
Diversity and Distribution of Marine Synechococcus: Multiple Gene Phylogenies for Consensus Classification and Development of qPCR Assays for Sensitive Measurement of Clades in the Ocean
Abstract
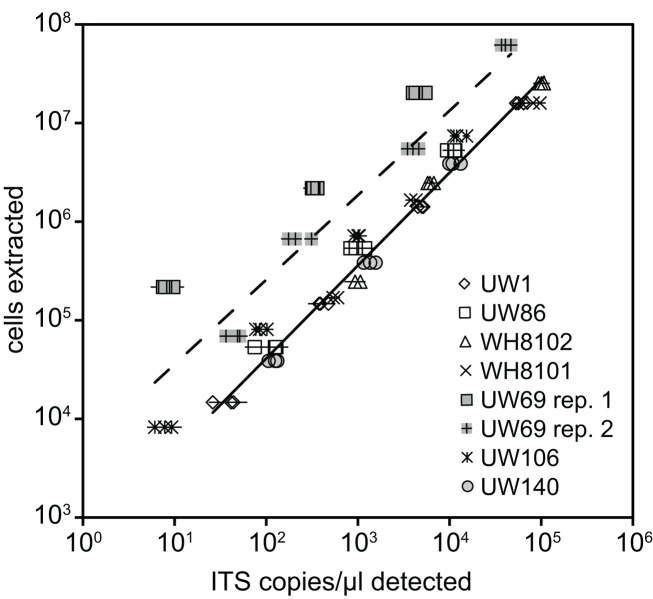
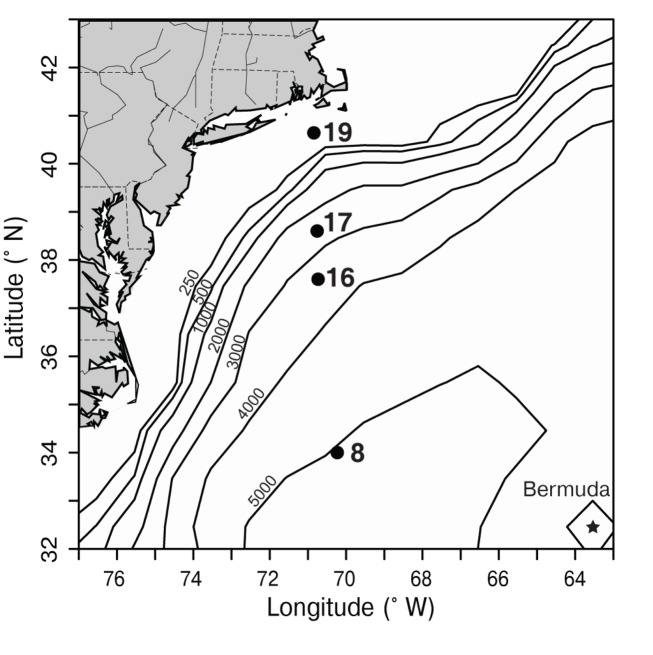
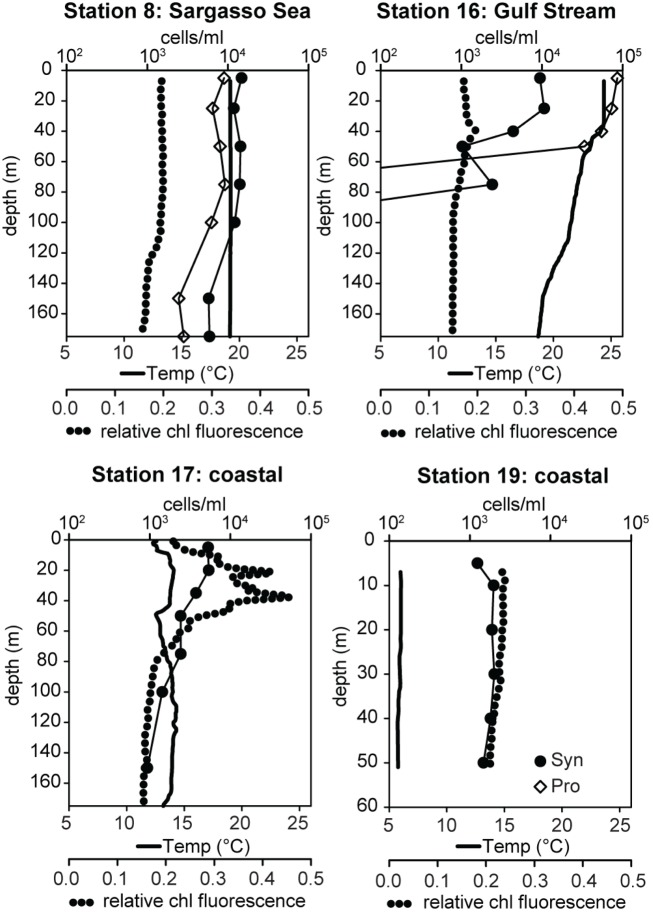
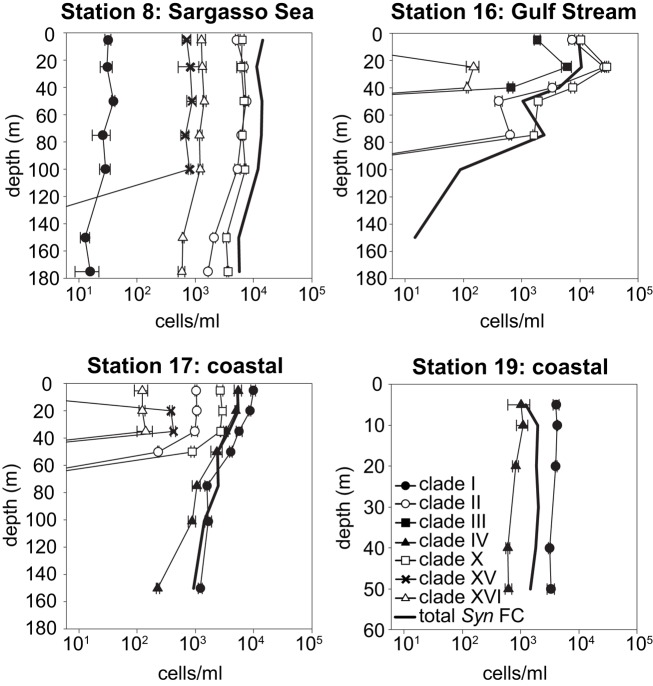
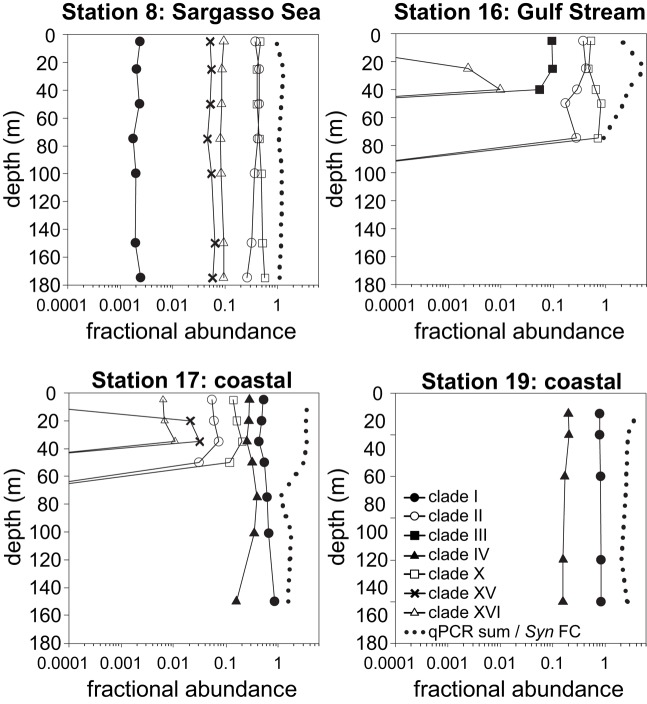
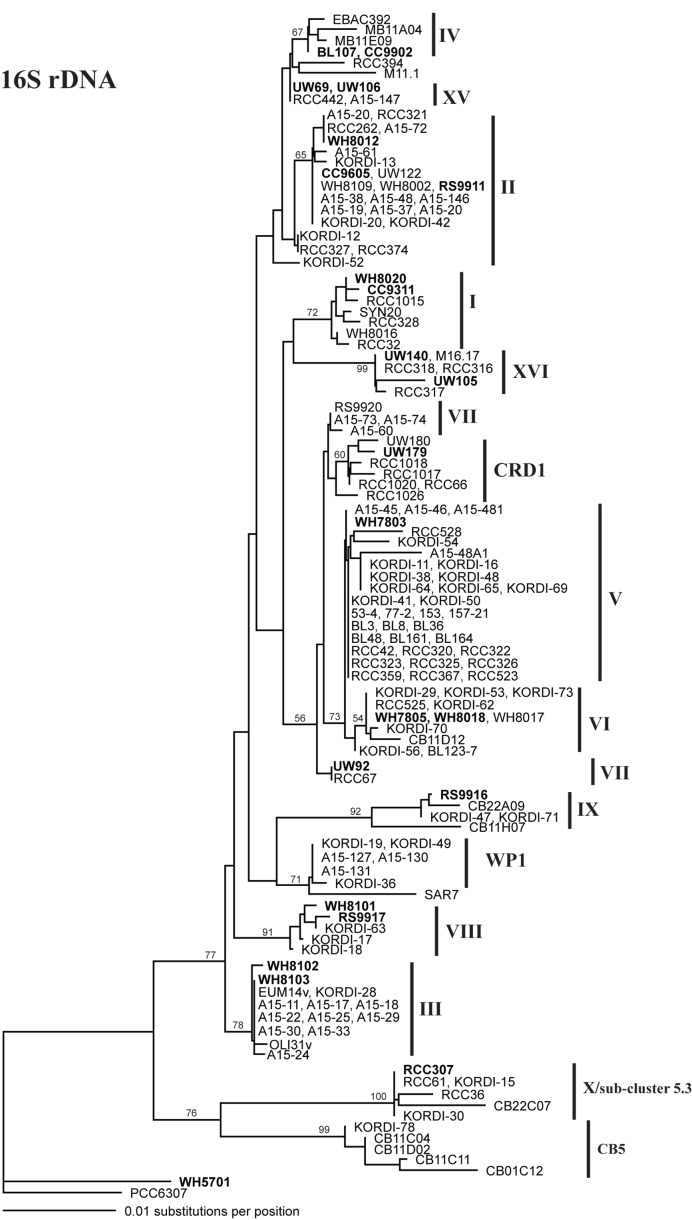
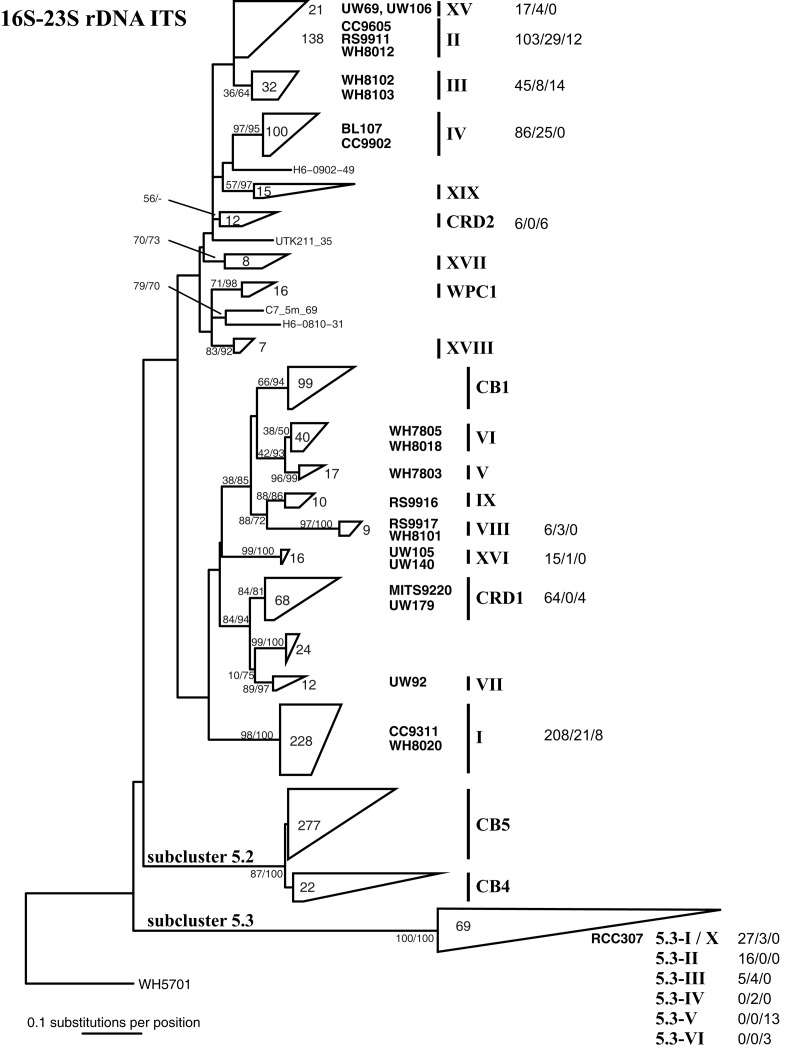
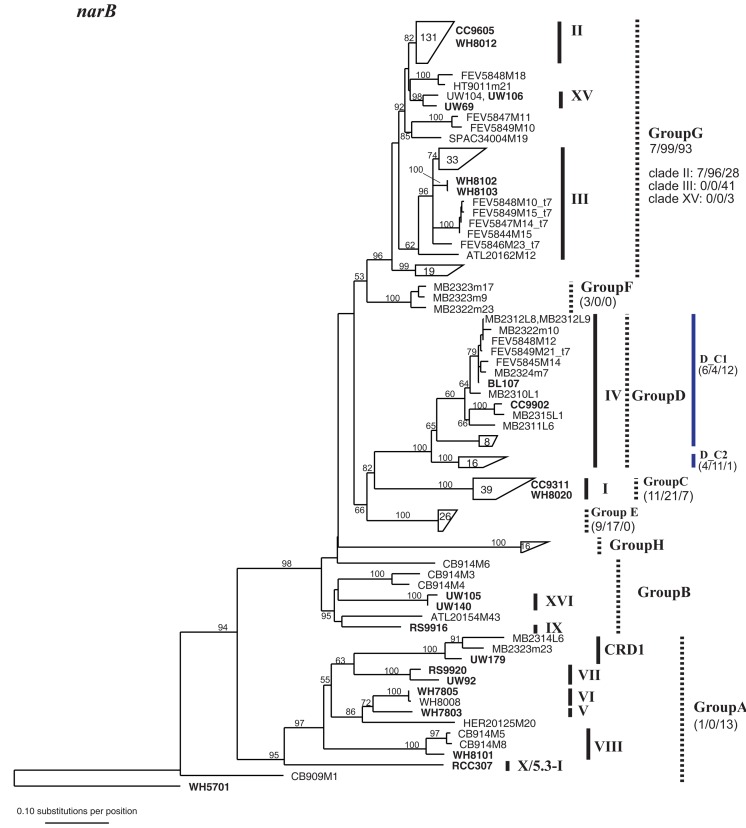
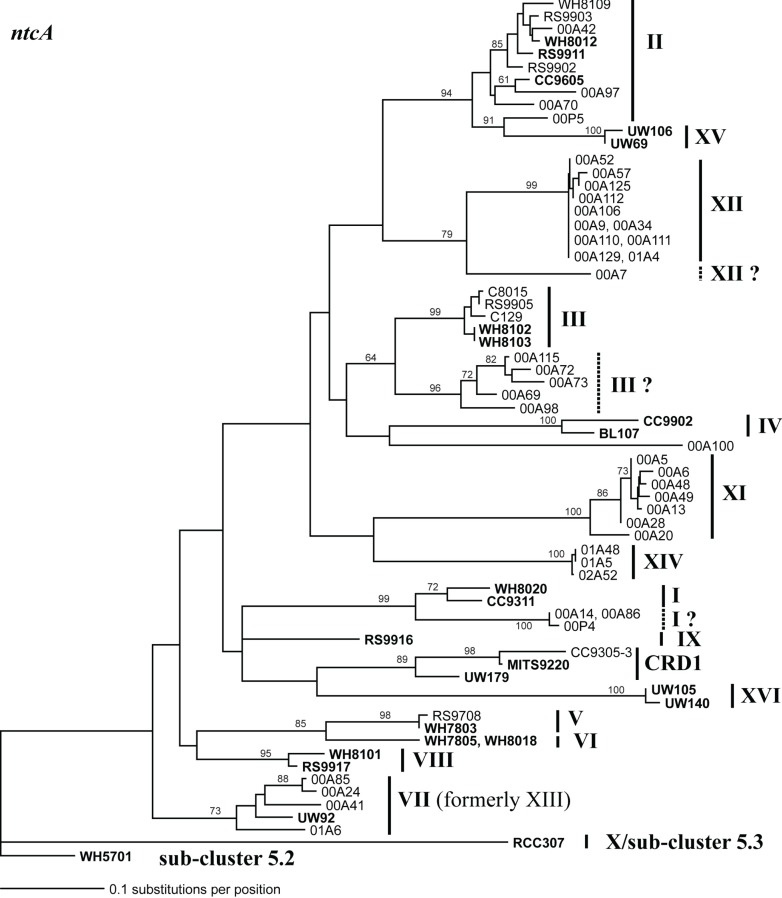
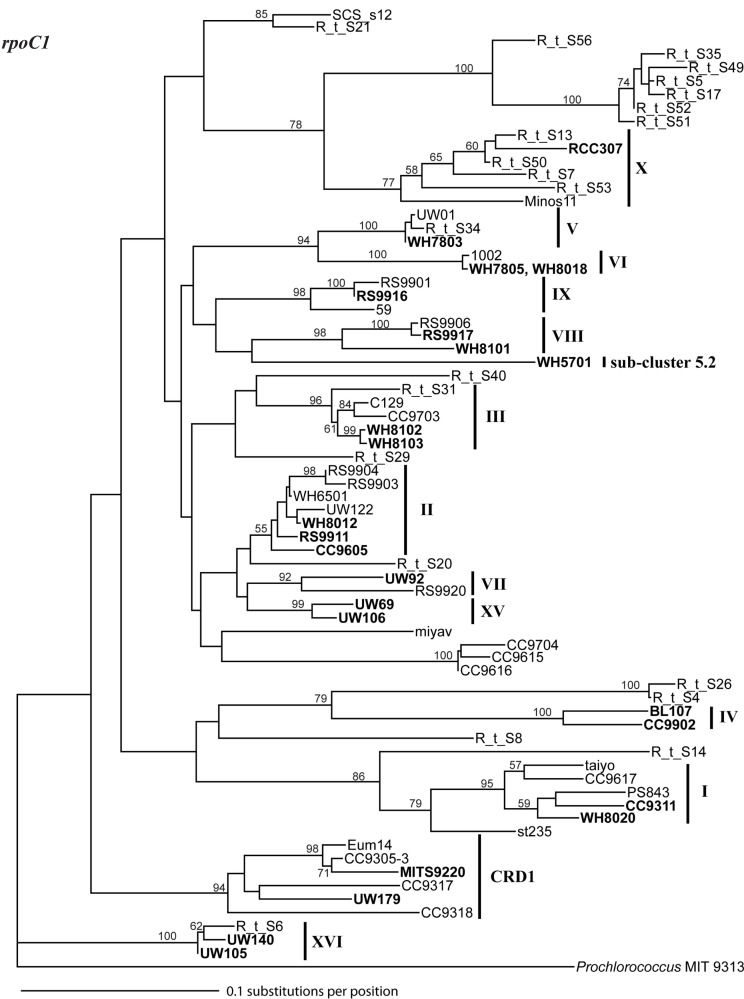
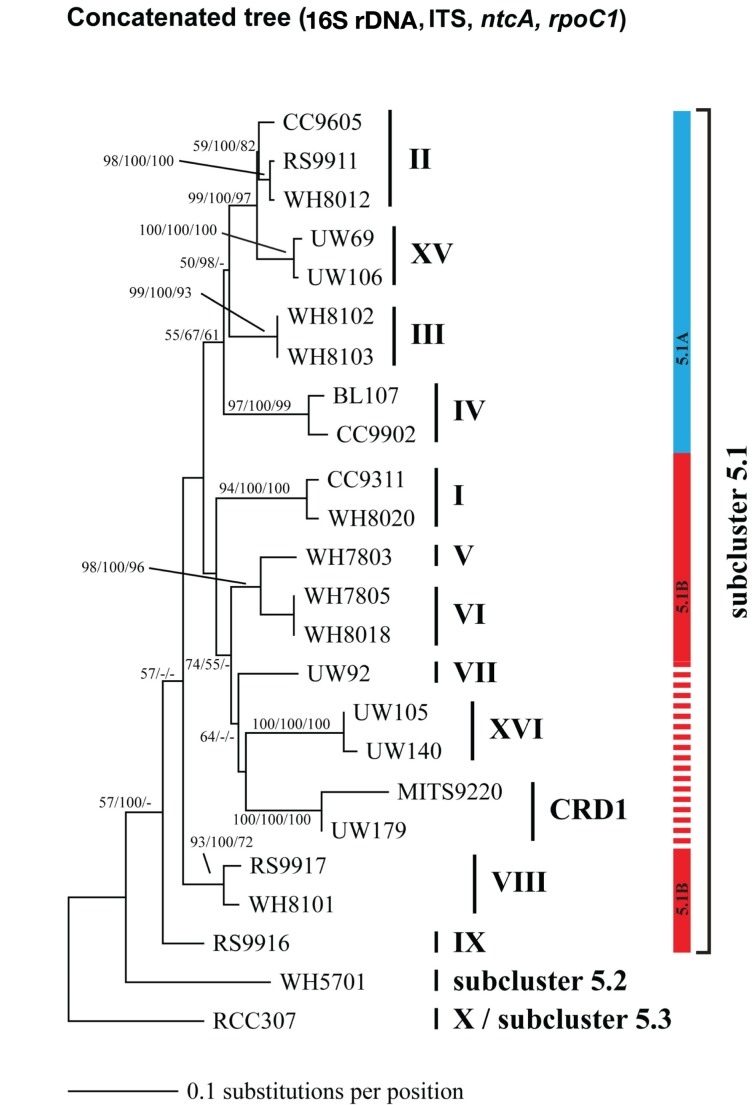
Marine Synechococcus is a globally significant genus of cyanobacteria that is comprised of multiple genetic lineages or clades. These clades are thought to represent ecologically distinct units, or ecotypes. Because multiple clades often co-occur together in the oceans, Synechococcus are ideal microbes to explore how closely related bacterial taxa within the same functional guild of organisms co-exist and partition marine habitats. Here we sequenced multiple gene loci from cultured strains to confirm the congruency of clade classifications between the 16S-23S rDNA internally transcribed spacer (ITS), 16S rDNA, narB, ntcA, and rpoC1 loci commonly used in Synechococcus diversity studies. We designed quantitative PCR (qPCR) assays that target the ITS for 10 Synechococcus clades, including four clades, XV, XVI, CRD1, and CRD2, not covered by previous assays employing other loci. Our new qPCR assays are very sensitive and specific, detecting down to tens of cells per ml. Application of these qPCR assays to field samples from the northwest Atlantic showed clear shifts in Synechococcus community composition across a coastal to open-ocean transect. Consistent with previous studies, clades I and IV dominated cold, coastal Synechococcus communities. Clades II and X were abundant at the two warmer, off-shore stations, and at all stations multiple Synechococcus clades co-occurred. qPCR assays developed here provide valuable tools to further explore the dynamics of microbial community structure and the mechanisms of co-existence.
Keywords: Synechococcus; biogeography; cyanobacteria; ecotype; microbial diversity; microbial ecology; multiple gene locus phylogeny; quantitative PCR.
Figures

References
-
- Beja O., Suzuki M. T., Koonin E. V., Aravind L., Hadd A., Nguyen L. P., Villacorta R., Amjadi M., Garrigues C., Jovanovich S. B., Feldman R. A., DeLong E. F. (2000). Construction and analysis of bacterial artificial chromosome libraries from a marine microbial assemblage. Environ. Microbiol. 2, 516–52910.1046/j.1462-2920.2000.00133.x - DOI - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases