

Estrogen modulates NFκB signaling by enhancing IκBα levels and blocking p65 binding at the promoters of inflammatory genes via estrogen receptor-β
- PMID: 22723832
- PMCID: PMC3378567
- DOI: 10.1371/journal.pone.0036890
Estrogen modulates NFκB signaling by enhancing IκBα levels and blocking p65 binding at the promoters of inflammatory genes via estrogen receptor-β
Abstract
Background: NFκB signaling is critical for expression of genes involved in the vascular injury response. We have shown that estrogen (17β-estradiol, E2) inhibits expression of these genes in an estrogen receptor (ER)-dependent manner in injured rat carotid arteries and in tumor necrosis factor (TNF)-α treated rat aortic smooth muscle cells (RASMCs). This study tested whether E2 inhibits NFκB signaling in RASMCs and defined the mechanisms.
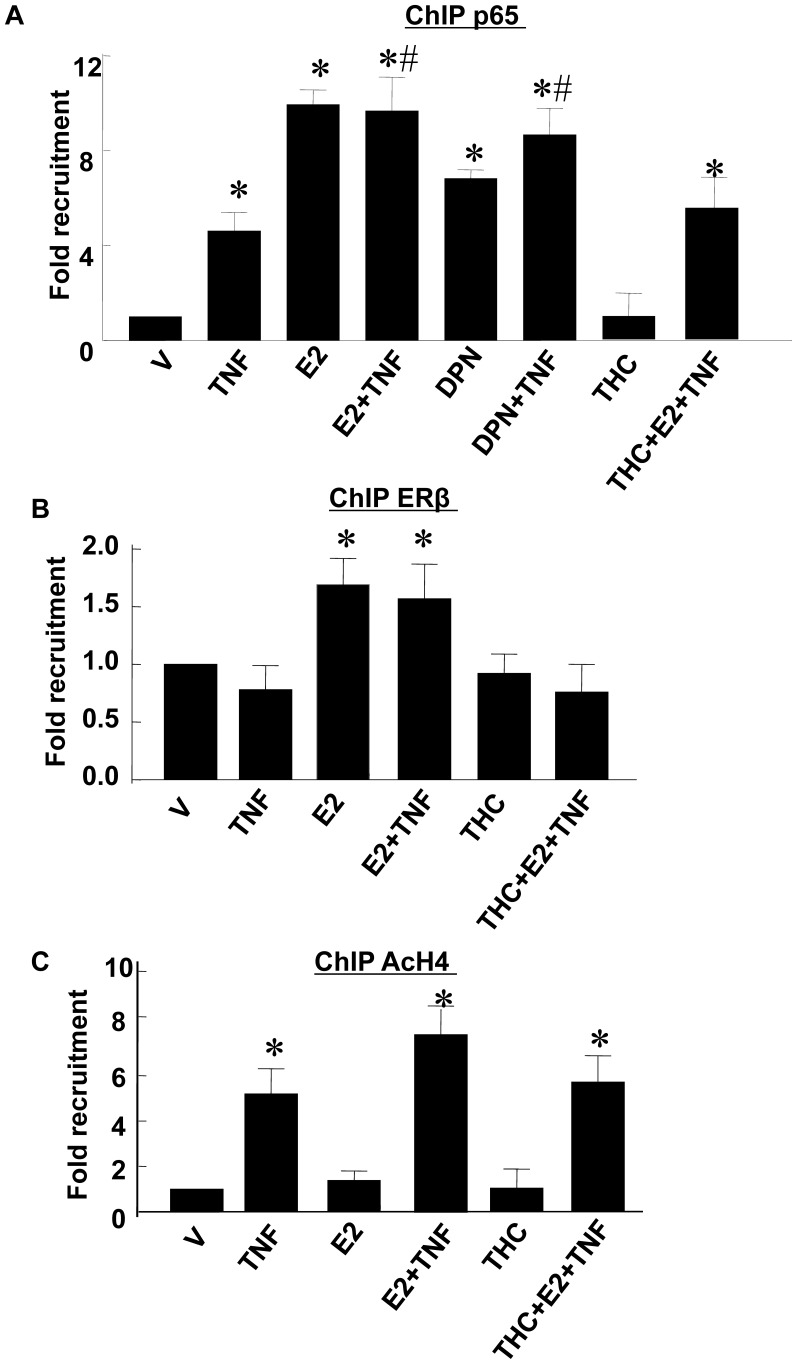
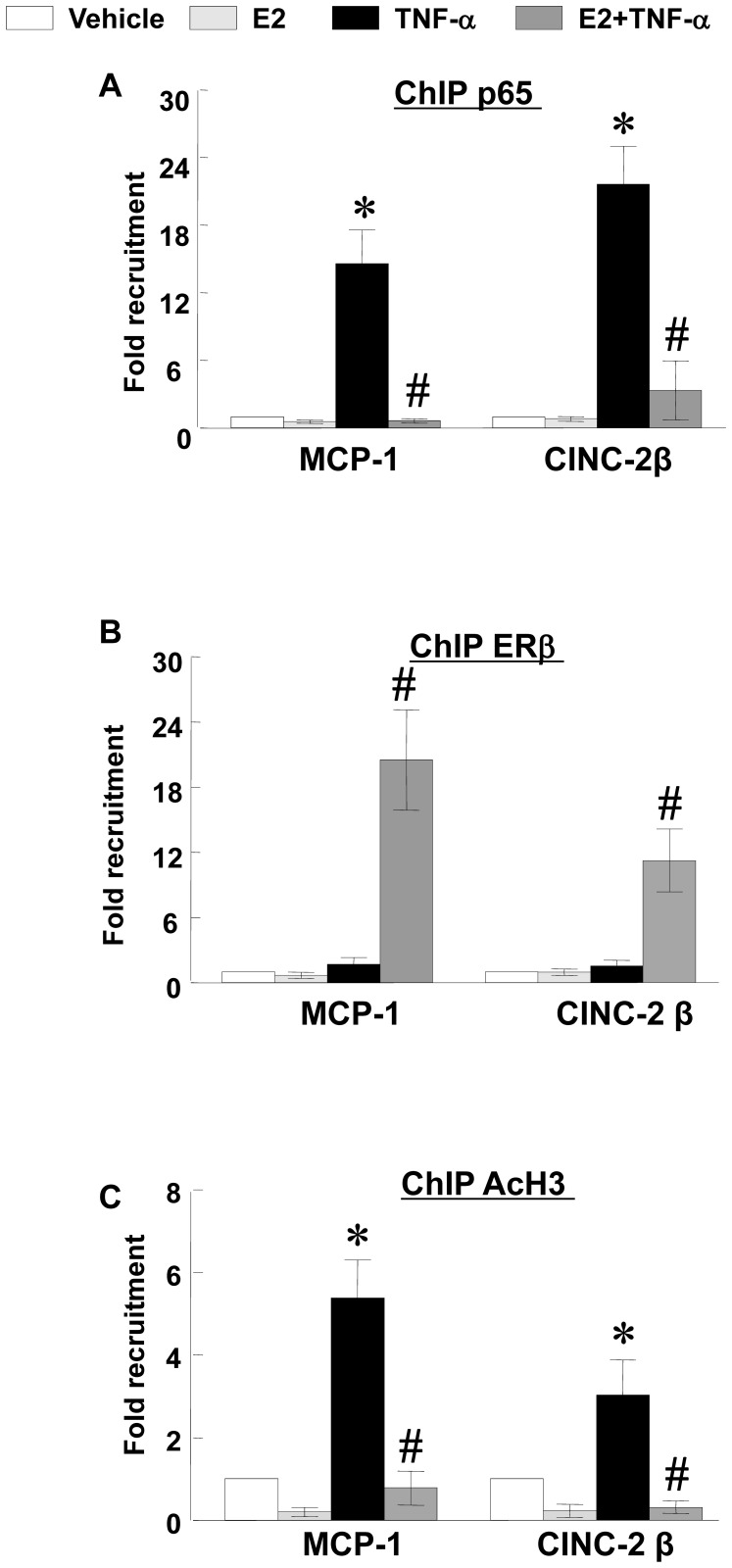
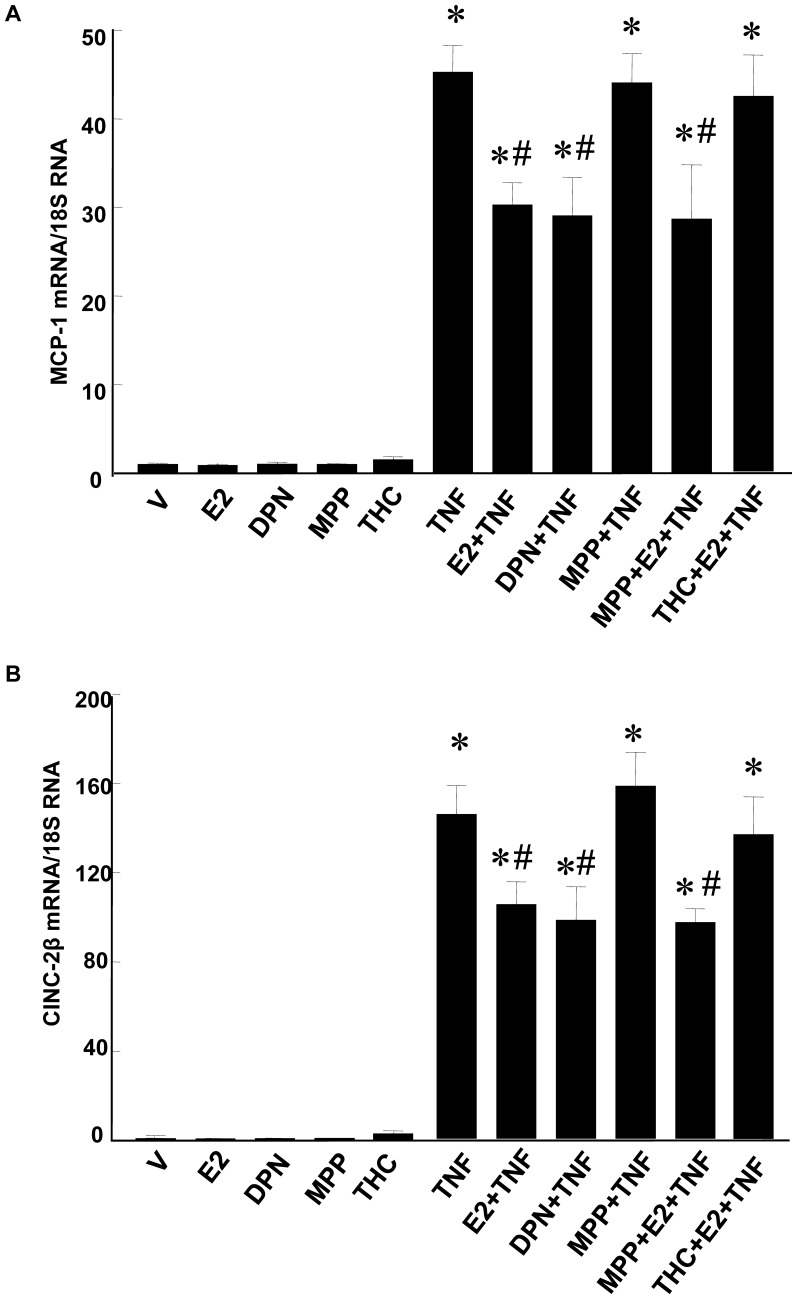
Methodology/principal findings: TNF-α treated RASMCs demonstrated rapid degradation of IκBα (10-30 min), followed by dramatic increases in IκBα mRNA and protein synthesis (40-60 min). E2 enhanced TNF-α induced IκBα synthesis without affecting IκBα degradation. Chromatin immunoprecipitation (ChIP) assays revealed that E2 pretreatment both enhanced TNF-α induced binding of NFκB p65 to the IκBα promoter and suppressed TNF-α induced binding of NFκB p65 to and reduced the levels of acetylated histone 3 at promoters of monocyte chemotactic protein (MCP)-1 and cytokine-induced neutrophil chemoattractant (CINC)-2β genes. ChIP analyses also demonstrated that ERβ can be recruited to the promoters of MCP-1 and CINC-2β during co-treatment with TNF-α and E2.
Conclusions: These data demonstrate that E2 inhibits inflammation in RASMCs by two distinct mechanisms: promoting new synthesis of IκBα, thus accelerating a negative feedback loop in NFκB signaling, and directly inhibiting binding of NFκB to the promoters of inflammatory genes. This first demonstration of multifaceted modulation of NFκB signaling by E2 may represent a novel mechanism by which E2 protects the vasculature against inflammatory injury.
Conflict of interest statement
Figures




Similar articles
-
Estrogen modulates TNF-alpha-induced inflammatory responses in rat aortic smooth muscle cells through estrogen receptor-beta activation.Am J Physiol Heart Circ Physiol. 2007 Jun;292(6):H2607-12. doi: 10.1152/ajpheart.01107.2006. Epub 2007 Jan 19. Am J Physiol Heart Circ Physiol. 2007. PMID: 17237237
-
O-GlcNAc modification of NFκB p65 inhibits TNF-α-induced inflammatory mediator expression in rat aortic smooth muscle cells.PLoS One. 2011;6(8):e24021. doi: 10.1371/journal.pone.0024021. Epub 2011 Aug 31. PLoS One. 2011. PMID: 21904602 Free PMC article.
-
The ERβ-CXCL19/CXCR4-NFκB pathway is critical in mediating the E2-induced inflammation response in the orange-spotted grouper (Epinephelus coioides).J Steroid Biochem Mol Biol. 2021 Sep;212:105926. doi: 10.1016/j.jsbmb.2021.105926. Epub 2021 Jun 4. J Steroid Biochem Mol Biol. 2021. PMID: 34091027
-
Estrogen, NFkappaB, and the heat shock response.Mol Med. 2008 Jul-Aug;14(7-8):517-27. doi: 10.2119/2008-00026.Stice. Mol Med. 2008. PMID: 18431462 Free PMC article. Review.
-
Role of disorder in IκB-NFκB interaction.IUBMB Life. 2012 Jun;64(6):499-505. doi: 10.1002/iub.1044. Epub 2012 May 9. IUBMB Life. 2012. PMID: 22573609 Free PMC article. Review.
Cited by
-
Hormonal regulation of the immune microenvironment in the mammary gland.J Mammary Gland Biol Neoplasia. 2014 Jul;19(2):229-39. doi: 10.1007/s10911-014-9324-x. Epub 2014 Jul 4. J Mammary Gland Biol Neoplasia. 2014. PMID: 24993978 Review.
-
The NF-κB-HE4 axis: A novel regulator of HE4 secretion in ovarian cancer.PLoS One. 2024 Dec 2;19(12):e0314564. doi: 10.1371/journal.pone.0314564. eCollection 2024. PLoS One. 2024. PMID: 39621651 Free PMC article.
-
Bone equilibria and disruptions.J Pediatr Soc North Am. 2024 Apr 6;7:100059. doi: 10.1016/j.jposna.2024.100059. eCollection 2024 May. J Pediatr Soc North Am. 2024. PMID: 40433272 Free PMC article.
-
Bortezomib and Arsenic Trioxide Activity on a Myelodysplastic Cell Line (P39): A Gene Expression Study.Turk J Haematol. 2015 Sep;32(3):206-12. doi: 10.4274/tjh.2014.0058. Turk J Haematol. 2015. PMID: 25913414 Free PMC article.
-
Dysfunction of Human Estrogen Signaling as a Novel Molecular Signature of Polycystic Ovary Syndrome.Int J Mol Sci. 2023 Nov 24;24(23):16689. doi: 10.3390/ijms242316689. Int J Mol Sci. 2023. PMID: 38069013 Free PMC article.
References
-
- Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115– 126. - PubMed
-
- Okamoto E, Couse T, De Leon H, Vinten-Johansen J, Goodman RB, et al. Perivascular inflammation after balloon angioplasty of porcine coronary arteries. Circulation. 2001;104:2228– 2235. - PubMed
-
- Buffon A, Biasucci LM, Liuzzo G, D’Onofrio G, Crea F, et al. Widespread coronary inflammation in unstable angina. N Engl J Med. 2002;347:5– 12. - PubMed
-
- Welt FG, Rogers C. Inflammation and restenosis in the stent era. Arterioscler Thromb Vasc Biol. 2002;22:1769– 1776. - PubMed
-
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868– 874. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL75211/HL/NHLBI NIH HHS/United States
- R01 HL087980/HL/NHLBI NIH HHS/United States
- HL044195/HL/NHLBI NIH HHS/United States
- R01 HL080017/HL/NHLBI NIH HHS/United States
- HL087980/HL/NHLBI NIH HHS/United States
- R01 HL075211/HL/NHLBI NIH HHS/United States
- P60 DK079626/DK/NIDDK NIH HHS/United States
- T35 HL007473/HL/NHLBI NIH HHS/United States
- P60 DK-079626/DK/NIDDK NIH HHS/United States
- HL080017/HL/NHLBI NIH HHS/United States
- T32 HL007457/HL/NHLBI NIH HHS/United States
- HL07457/HL/NHLBI NIH HHS/United States
- R01 HL044195/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
