

TNFR1-dependent pulmonary apoptosis during ischemic acute kidney injury
- PMID: 22728466
- PMCID: PMC3468427
- DOI: 10.1152/ajplung.00301.2011
TNFR1-dependent pulmonary apoptosis during ischemic acute kidney injury
Abstract
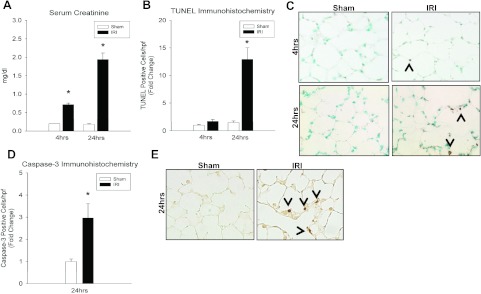
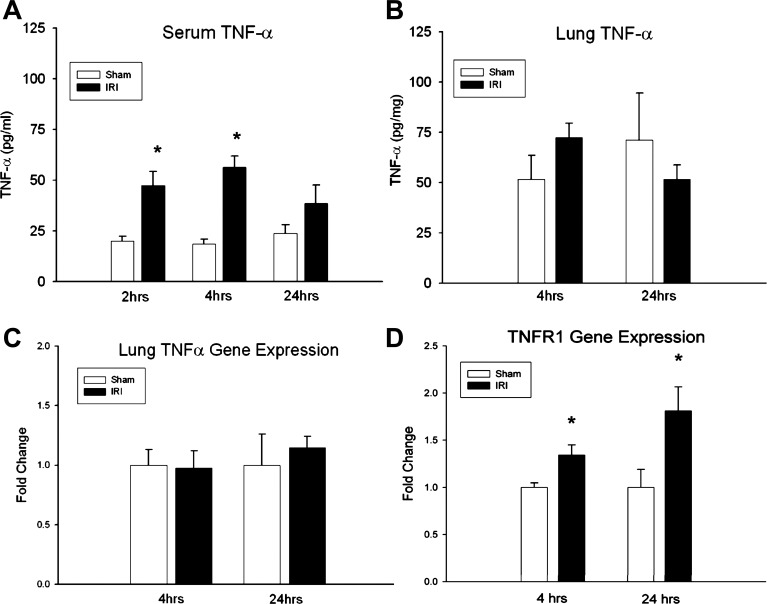
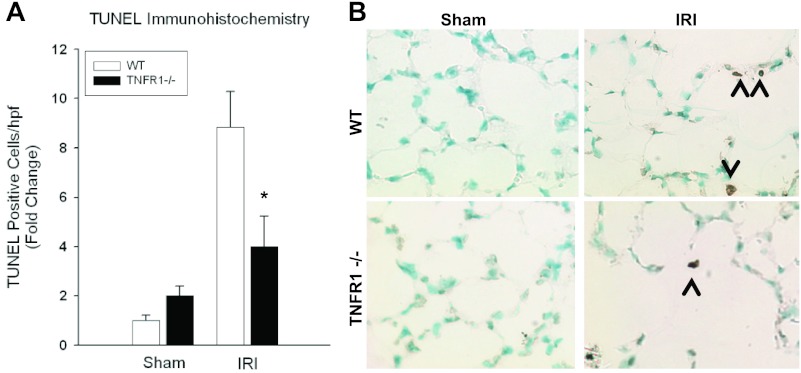
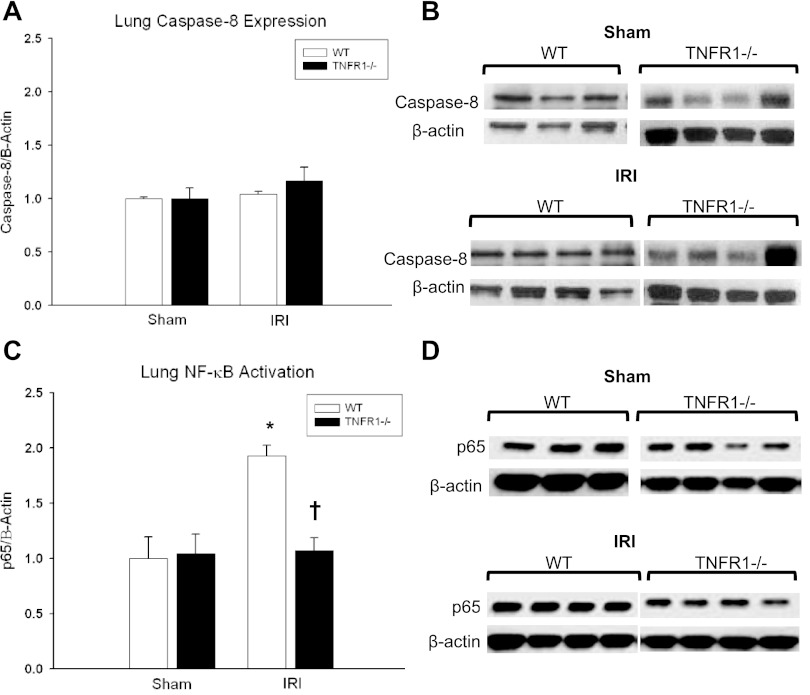
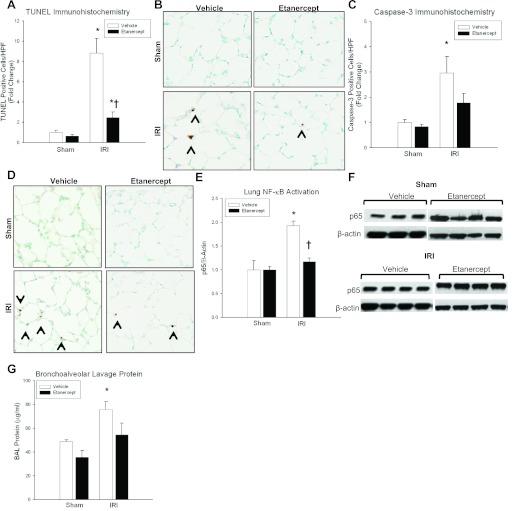
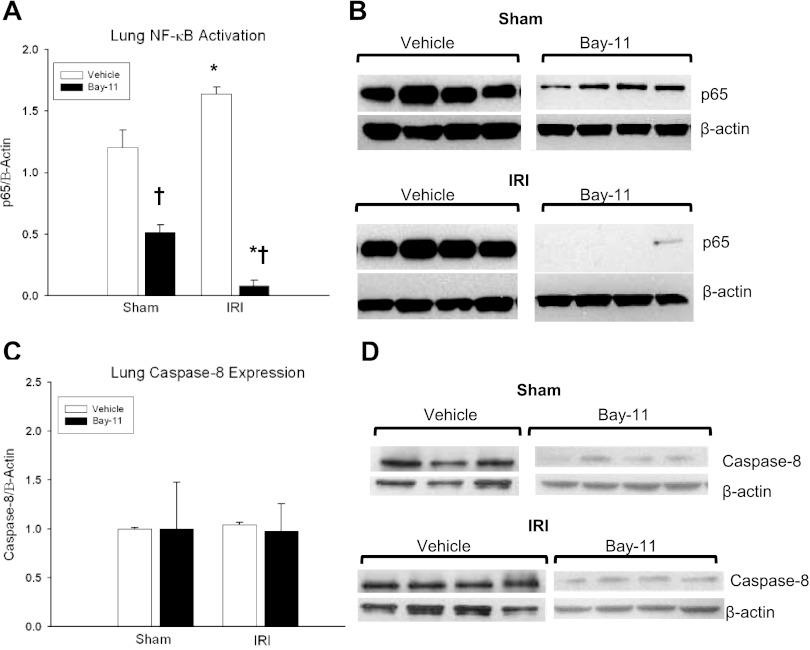
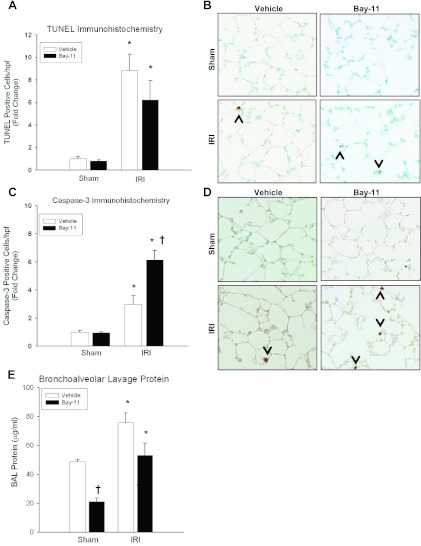
Despite advancements in renal replacement therapy, the mortality rate for acute kidney injury (AKI) remains unacceptably high, likely due to remote organ injury. Kidney ischemia-reperfusion injury (IRI) activates cellular and soluble mediators that incite a distinct pulmonary proinflammatory and proapoptotic response. Tumor necrosis factor receptor 1 (TNFR1) has been identified as a prominent death receptor activated in the lungs during ischemic AKI. We hypothesized that circulating TNF-α released from the postischemic kidney induces TNFR1-mediated pulmonary apoptosis, and we aimed to elucidate molecular pathways to programmed cell death. Using an established murine model of kidney IRI, we characterized the time course for increased circulatory and pulmonary TNF-α levels and measured concurrent upregulation of pulmonary TNFR1 expression. We then identified TNFR1-dependent pulmonary apoptosis after ischemic AKI using TNFR1-/- mice. Subsequent TNF-α signaling disruption with Etanercept implicated circulatory TNF-α as a key soluble mediator of pulmonary apoptosis and lung microvascular barrier dysfunction during ischemic AKI. We further elucidated pathways of TNFR1-mediated apoptosis with NF-κB (Complex I) and caspase-8 (Complex II) expression and discovered that TNFR1 proapoptotic signaling induces NF-κB activation. Additionally, inhibition of NF-κB (Complex I) resulted in a proapoptotic phenotype, lung barrier leak, and altered cellular flice inhibitory protein signaling independent of caspase-8 (Complex II) activation. Ischemic AKI activates soluble TNF-α and induces TNFR1-dependent pulmonary apoptosis through augmentation of the prosurvival and proapoptotic TNFR1 signaling pathway. Kidney-lung crosstalk after ischemic AKI represents a complex pathological process, yet focusing on specific biological pathways may yield potential future therapeutic targets.
Figures
References
-
- Cataisson C, Pearson AJ, Torgerson S, Nedospasov SA, Yuspa SH. Protein kinase C alpha-mediated chemotaxis of neutrophils requires NF-kappa B activity but is independent of TNF alpha signaling in mouse skin in vivo. J Immunol 174: 1686–1692, 2005 - PubMed
-
- Donnahoo KK, Meng X, Ayala A, Cain MP, Harken AH, Meldrum DR. Early kidney TNF-α expression mediates neutrophil infiltration and injury after renal ischemia-reperfusion. Am J Physiol Regul Integr Comp Physiol 277: R922–R929, 1999 - PubMed
-
- Geering B, Gurzeler U, Federzoni E, Kaufmann T, Simon HU. A novel TNFR1-triggered apoptosis pathway mediated by class IA PI3Ks in neutrophils. Blood 117: 5953–5962, 2011 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases