

PDLIM2 restricts Th1 and Th17 differentiation and prevents autoimmune disease
- PMID: 22731402
- PMCID: PMC3543335
- DOI: 10.1186/2045-3701-2-23
PDLIM2 restricts Th1 and Th17 differentiation and prevents autoimmune disease
Abstract
Background: PDLIM2 is essential for the termination of the inflammatory transcription factors NF-κB and STAT but is dispensable for the development of immune cells and immune tissues/organs. Currently, it remains unknown whether and how PDLIM2 is involved in physiologic and pathogenic processes.
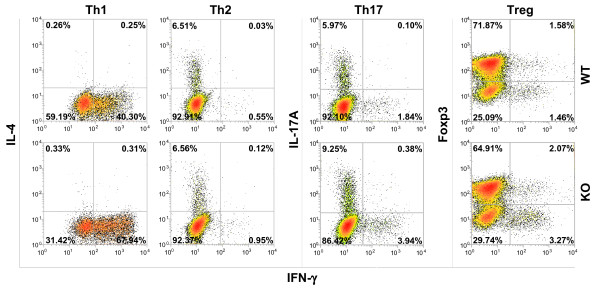
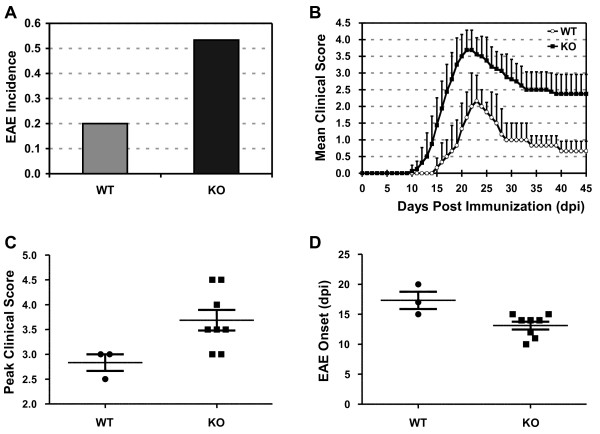
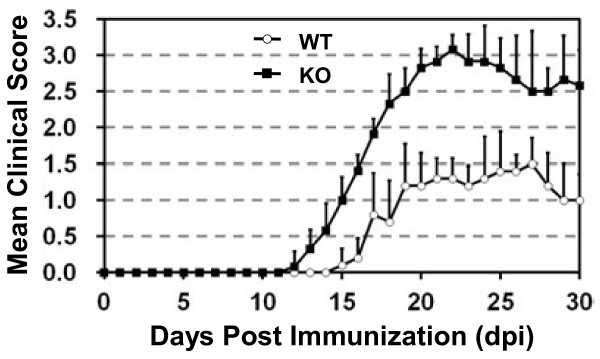
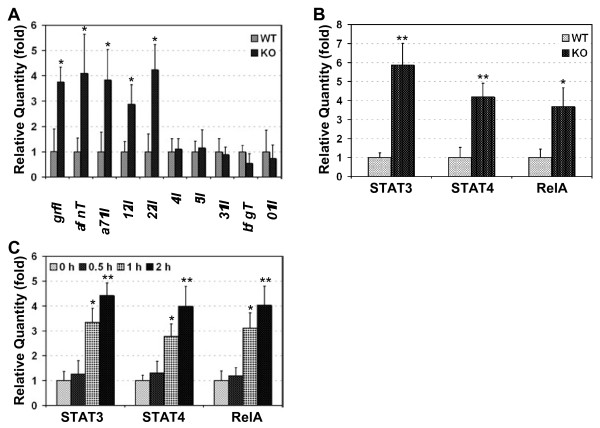
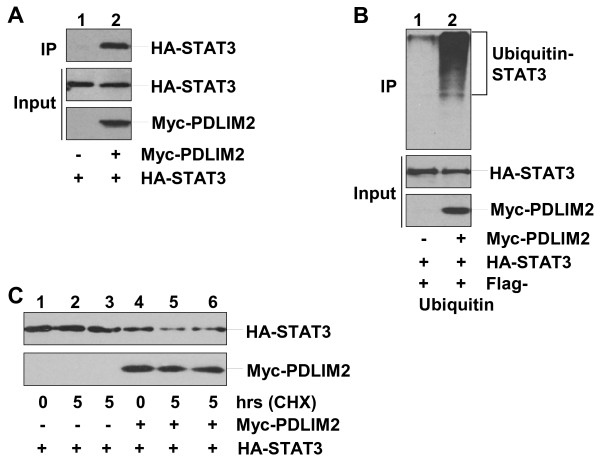
Results: Here we report that naive PDLIM2 deficient CD4+ T cells were prone to differentiate into Th1 and Th17 cells. PDLIM2 deficiency, however, had no obvious effect on lineage commitment towards Th2 or Treg cells. Notably, PDLIM2 deficient mice exhibited increased susceptibility to experimental autoimmune encephalitis (EAE), a Th1 and/or Th17 cell-mediated inflammatory disease model of multiple sclerosis (MS). Mechanistic studies further indicate that PDLIM2 was required for restricting expression of Th1 and Th17 cytokines, which was in accordance with the role of PDLIM2 in the termination of NF-κB and STAT activation.
Conclusion: These findings suggest that PDLIM2 is a key modulator of T-cell-mediated immune responses that may be targeted for the therapy of human autoimmune diseases.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
