

doi: 10.1186/gb-2012-13-6-r56.
Toward almost closed genomes with GapFiller
Affiliations
- PMID: 22731987
- PMCID: PMC3446322
- DOI: 10.1186/gb-2012-13-6-r56
Item in Clipboard
Toward almost closed genomes with GapFiller
Genome Biol.
.
Abstract
De novo assembly is a commonly used application of next-generation sequencing experiments. The ultimate goal is to puzzle millions of reads into one complete genome, although draft assemblies usually result in a number of gapped scaffold sequences. In this paper we propose an automated strategy, called GapFiller, to reliably close gaps within scaffolds using paired reads. The method shows good results on both bacterial and eukaryotic datasets, allowing only few errors. As a consequence, the amount of additional wetlab work needed to close a genome is drastically reduced. The software is available at http://www.baseclear.com/bioinformatics-tools/.
Figures
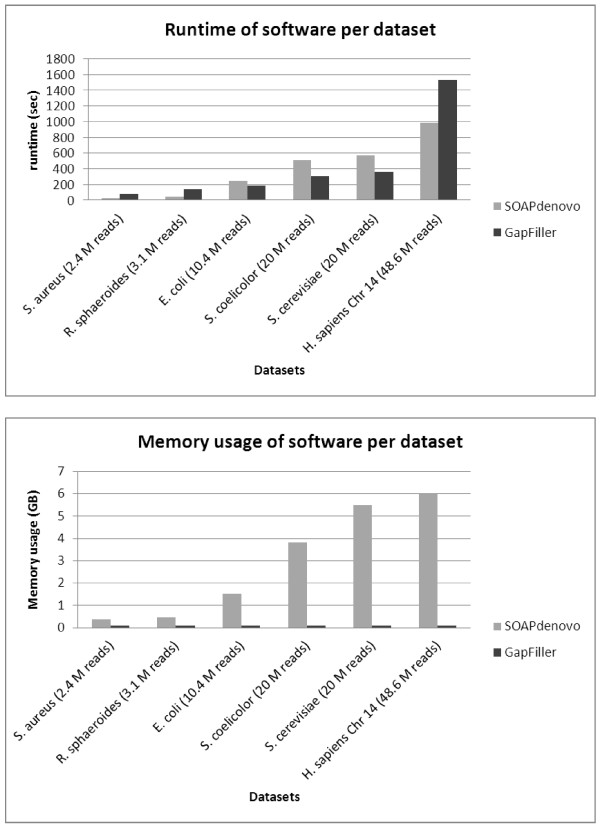
Time and memory consumption of gap closure software. Comparative analysis of the runtime and memory usage per dataset based on a single iteration. SOAPdenovo needs a shorter time to complete the analysis if the amount of data is very small or large, whereas GapFiller is faster for intermediate data sizes (10 to 20 million reads). With regard to memory usage, GapFiller outperforms SOAPdenovo since intermediate output is temporarily stored (and not kept in the memory). For all datasets analyzed, GapFiller requires only 0.1 GB of memory, which is mostly consumed by the Burrows-Wheeler Aligner (BWA). Note that no results are displayed for IMAGE since the method can not handle multiple libraries and requires very large computation times to complete the process. M, million.
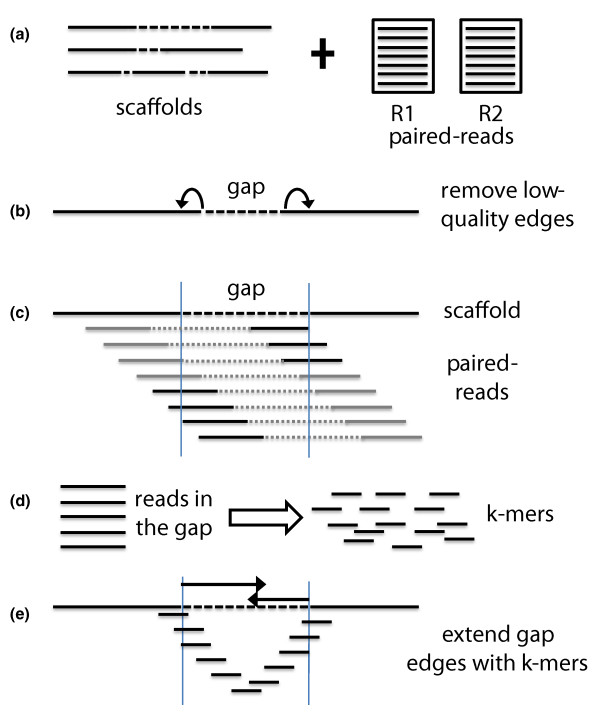
Schematic overview of the GapFiller algorithm. (a) The input data consist of a set of scaffold sequences containing gapped nucleotides and one or more sets of paired-end and/or mate-pair reads. (b) As a pre-processing step low quality nucleotides are removed from the sequence edges, thus enlarging the gap of ten nucleotides from each side. It should be stressed that the contig ends resulting from a draft assembly often contain misassemblies. (c) Paired-reads are aligned to the scaffolds and retained if one pair aligns to a scaffold sequence (dark grey) and one pair to a gapped region (black). (d) All pairs that are estimated to fall in the gapped regions are split into k-mers and used for gap filling. (e) The gap is closed from each edge by using k-mers that present a sequence overlap of size (k-mer - 1) and one nucleotide overhang. Gaps are closed if the right and left extensions can be merged and correspond to the estimated sequence gap.
References
-
- Li R, Fan W, Tian G, Zhu H, He L, Cai J, Huang Q, Cai Q, Li B, Bai Y, Zhang Z, Zhang Y, Wang W, Li J, Wei F, Li H, Jian M, Li J, Zhang Z, Nielsen R, Li D, Gu W, Yang Z, Xuan Z, Ryder OA, Leung FC, Zhou Y, Cao J, Sun X, Fu Y. et al. The sequence and de novo assembly of the giant panda genome. Nature. 2010;463:311–317. doi: 10.1038/nature08696. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
