

Advances in understanding the molecular basis of frontotemporal dementia
- PMID: 22732773
- PMCID: PMC3629543
- DOI: 10.1038/nrneurol.2012.117
Advances in understanding the molecular basis of frontotemporal dementia
Erratum in
- Nat Rev Neurol. 2013 May;9(5):240
Abstract
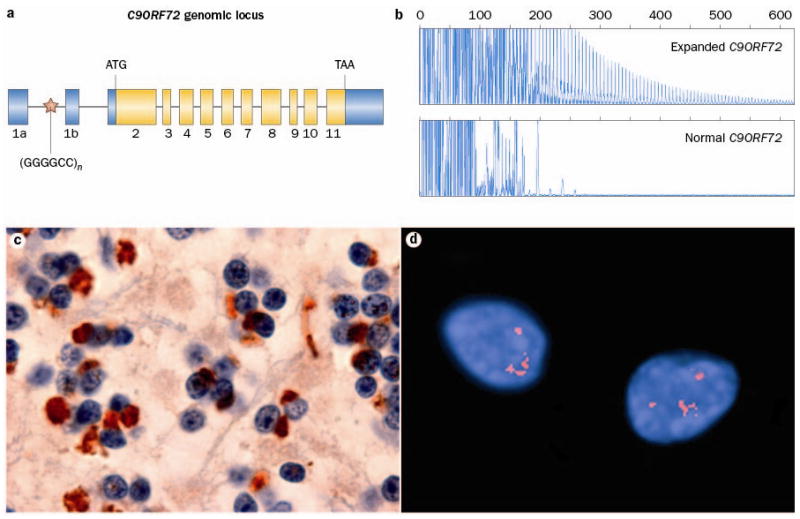
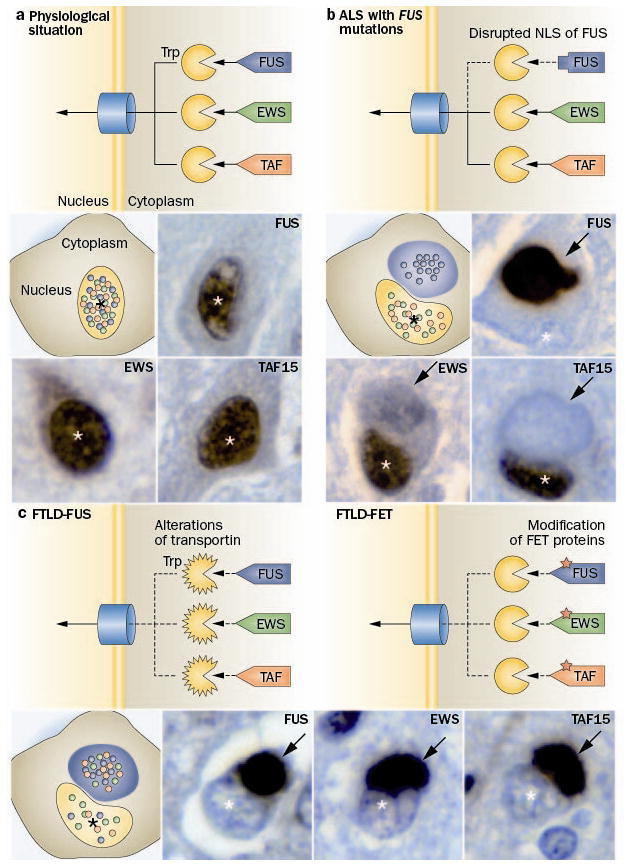
Frontotemporal dementia (FTD) is a clinical syndrome with a heterogeneous molecular basis. Until recently, the underlying cause was known in only a minority of cases that were associated with abnormalities of the tau protein or gene. In 2006, however, mutations in the progranulin gene were discovered as another important cause of familial FTD. That same year, TAR DNA-binding protein 43 (TDP-43) was identified as the pathological protein in the most common subtypes of FTD and amyotrophic lateral sclerosis (ALS). Since then, substantial efforts have been made to understand the functions and regulation of progranulin and TDP-43, as well as their roles in neurodegeneration. More recently, other DNA/RNA binding proteins (FET family proteins) have been identified as the pathological proteins in most of the remaining cases of FTD. In 2011, abnormal expansion of a hexanucleotide repeat in the gene C9orf72 was found to be the most common genetic cause of both FTD and ALS. All common FTD-causing genes have seemingly now been discovered and the main pathological proteins identified. In this Review, we highlight recent advances in understanding the molecular aspects of FTD, which will provide the basis for improved patient care through the development of more-targeted diagnostic tests and therapies.
Figures
References
-
- Bird T, et al. Epidemiology and genetics of frontotemporal dementia/Pick’s disease. Ann Neurol. 2003;54 (Suppl 5):S29–31. - PubMed
-
- Feldman H, et al. A Canadian cohort study of cognitive impairment and related dementias (ACCORD): study methods and baseline results. Neuroepidemiology. 2003;22:265–274. - PubMed
-
- McKhann GM, et al. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol. 2001;58:1803–1809. - PubMed
-
- Neary D, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
