

Critical role for calcium mobilization in activation of the NLRP3 inflammasome
- PMID: 22733741
- PMCID: PMC3396518
- DOI: 10.1073/pnas.1117765109
Critical role for calcium mobilization in activation of the NLRP3 inflammasome
Abstract
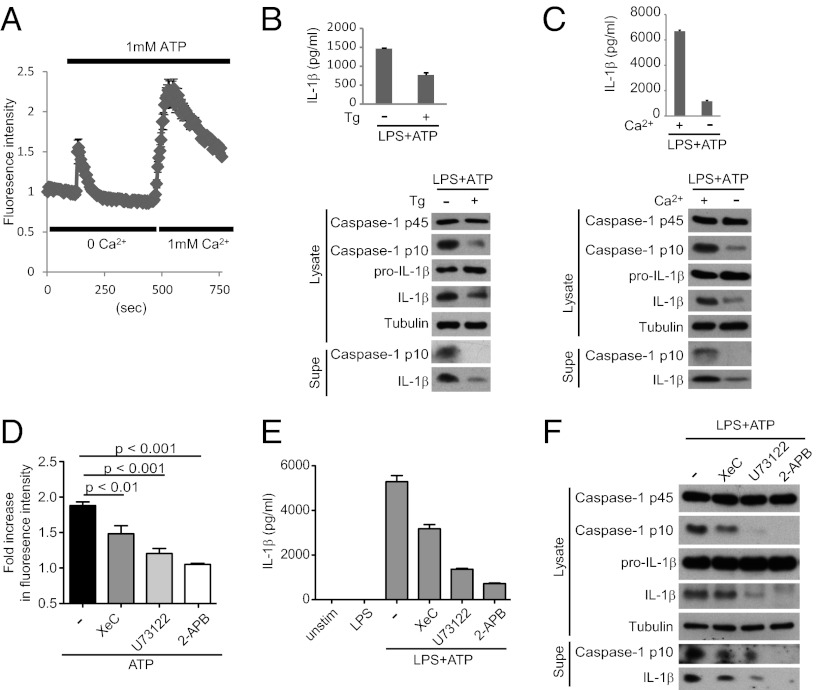
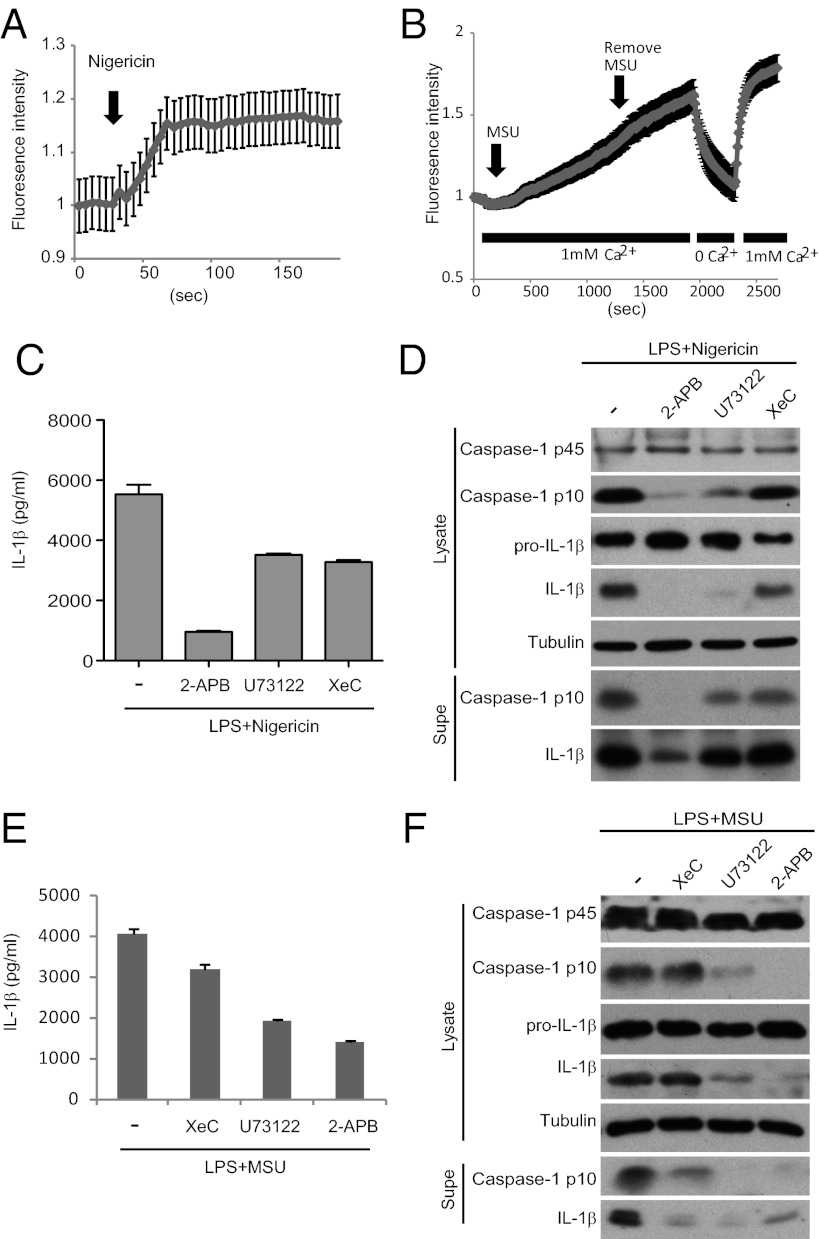
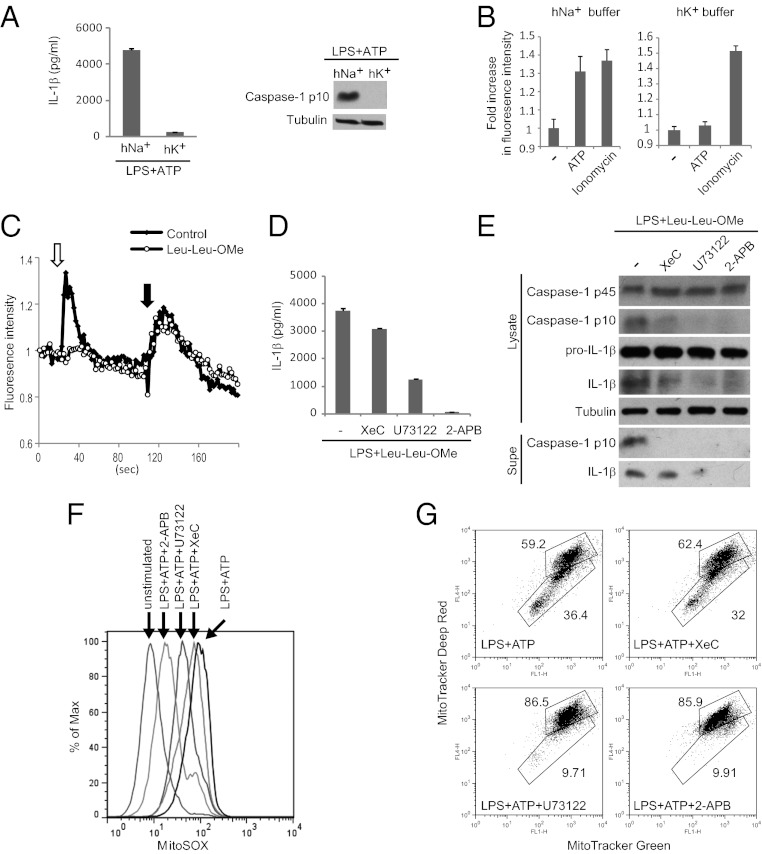
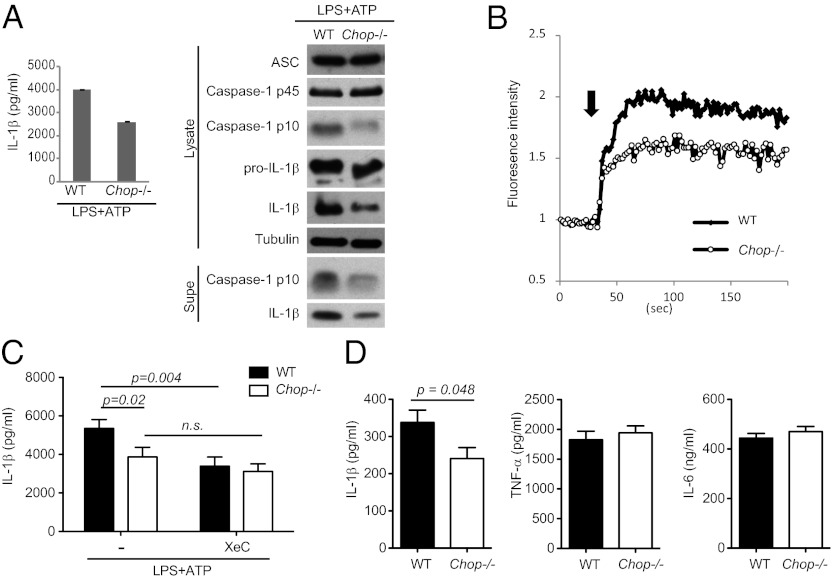
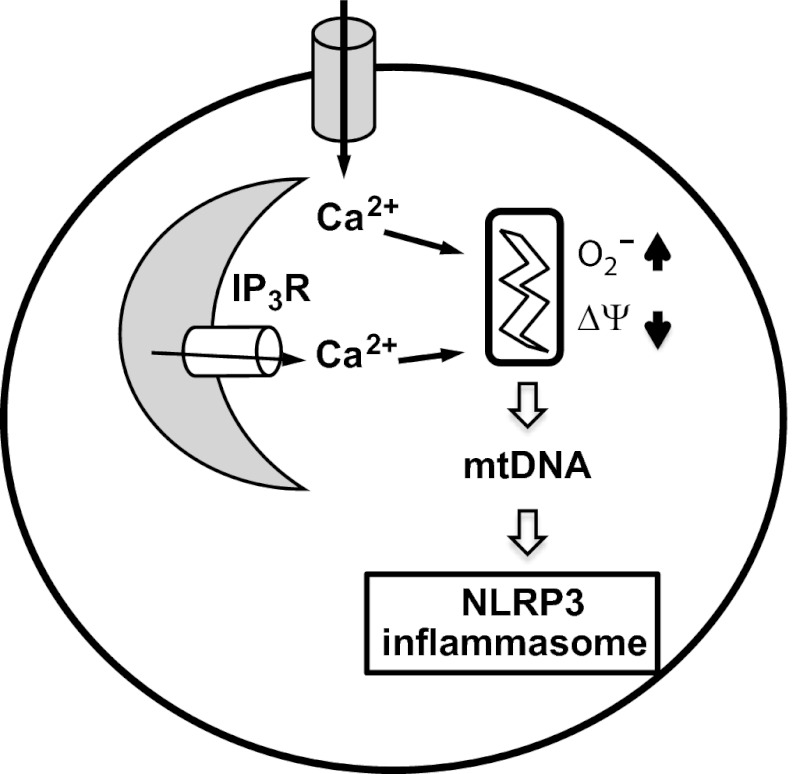
The NLRP3 (nucleotide-binding domain, leucine-rich-repeat-containing family, pyrin domain-containing 3) inflammasome mediates production of inflammatory mediators, such as IL-1β and IL-18, and as such is implicated in a variety of inflammatory processes, including infection, sepsis, autoinflammatory diseases, and metabolic diseases. The proximal steps in NLRP3 inflammasome activation are not well understood. Here we elucidate a critical role for Ca(2+) mobilization in activation of the NLRP3 inflammasome by multiple stimuli. We demonstrate that blocking Ca(2+) mobilization inhibits assembly and activation of the NLRP3 inflammasome complex, and that during ATP stimulation Ca(2+) signaling is pivotal in promoting mitochondrial damage. C/EPB homologous protein, a transcription factor that can modulate Ca(2+) release from the endoplasmic reticulum, amplifies NLRP3 inflammasome activation, thus linking endoplasmic reticulum stress to activation of the NLRP3 inflammasome. Our findings support a model for NLRP3 inflammasome activation by Ca(2+)-mediated mitochondrial damage.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
The Roles of Endoplasmic Reticulum in NLRP3 Inflammasome Activation.Cells. 2020 May 14;9(5):1219. doi: 10.3390/cells9051219. Cells. 2020. PMID: 32423023 Free PMC article. Review.
-
The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP.Nature. 2012 Dec 6;492(7427):123-7. doi: 10.1038/nature11588. Epub 2012 Nov 11. Nature. 2012. PMID: 23143333 Free PMC article.
-
Anthocyanins from Hibiscus syriacus L. Inhibit NLRP3 Inflammasome in BV2 Microglia Cells by Alleviating NF-κB- and ER Stress-Induced Ca2+ Accumulation and Mitochondrial ROS Production.Oxid Med Cell Longev. 2021 Feb 4;2021:1246491. doi: 10.1155/2021/1246491. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 33613822 Free PMC article.
-
Resveratrol inhibits the acetylated α-tubulin-mediated assembly of the NLRP3-inflammasome.Int Immunol. 2015 Sep;27(9):425-34. doi: 10.1093/intimm/dxv018. Epub 2015 Apr 7. Int Immunol. 2015. PMID: 25855661
-
Regulation and Function of the Nucleotide Binding Domain Leucine-Rich Repeat-Containing Receptor, Pyrin Domain-Containing-3 Inflammasome in Lung Disease.Am J Respir Cell Mol Biol. 2016 Feb;54(2):151-60. doi: 10.1165/rcmb.2015-0231TR. Am J Respir Cell Mol Biol. 2016. PMID: 26418144 Free PMC article. Review.
Cited by
-
Recent Advances of the NLRP3 Inflammasome in Central Nervous System Disorders.J Immunol Res. 2016;2016:9238290. doi: 10.1155/2016/9238290. Epub 2016 Aug 29. J Immunol Res. 2016. PMID: 27652274 Free PMC article. Review.
-
Relevant mediators involved in and therapies targeting the inflammatory response induced by activation of the NLRP3 inflammasome in ischemic stroke.J Neuroinflammation. 2021 May 31;18(1):123. doi: 10.1186/s12974-021-02137-8. J Neuroinflammation. 2021. PMID: 34059091 Free PMC article. Review.
-
NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: roles for K+ efflux and Ca2+ influx.Am J Physiol Cell Physiol. 2016 Jul 1;311(1):C83-C100. doi: 10.1152/ajpcell.00298.2015. Epub 2016 May 11. Am J Physiol Cell Physiol. 2016. PMID: 27170638 Free PMC article.
-
Ageing and Low-Level Chronic Inflammation: The Role of the Biological Clock.Antioxidants (Basel). 2022 Nov 11;11(11):2228. doi: 10.3390/antiox11112228. Antioxidants (Basel). 2022. PMID: 36421414 Free PMC article. Review.
-
Updated insights into the NLRP3 inflammasome in postoperative cognitive dysfunction: emerging mechanisms and treatments.Front Aging Neurosci. 2024 Sep 30;16:1480502. doi: 10.3389/fnagi.2024.1480502. eCollection 2024. Front Aging Neurosci. 2024. PMID: 39411285 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
