

IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells
- PMID: 22733744
- PMCID: PMC3411948
- DOI: 10.1073/pnas.1203692109
IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells
Abstract
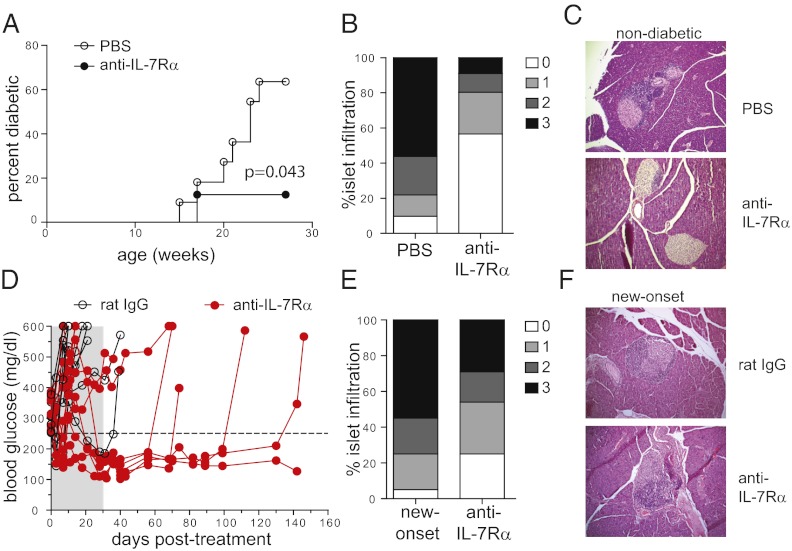
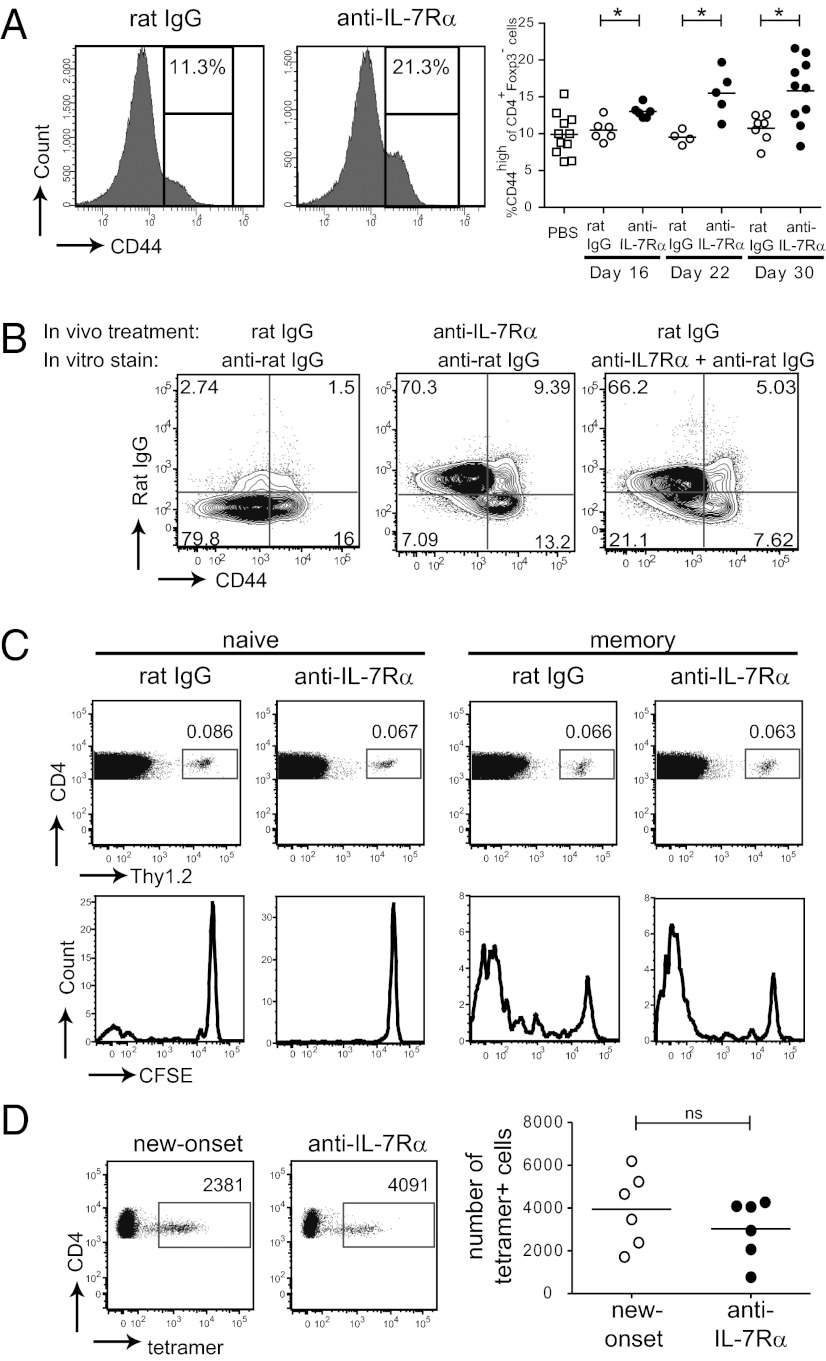
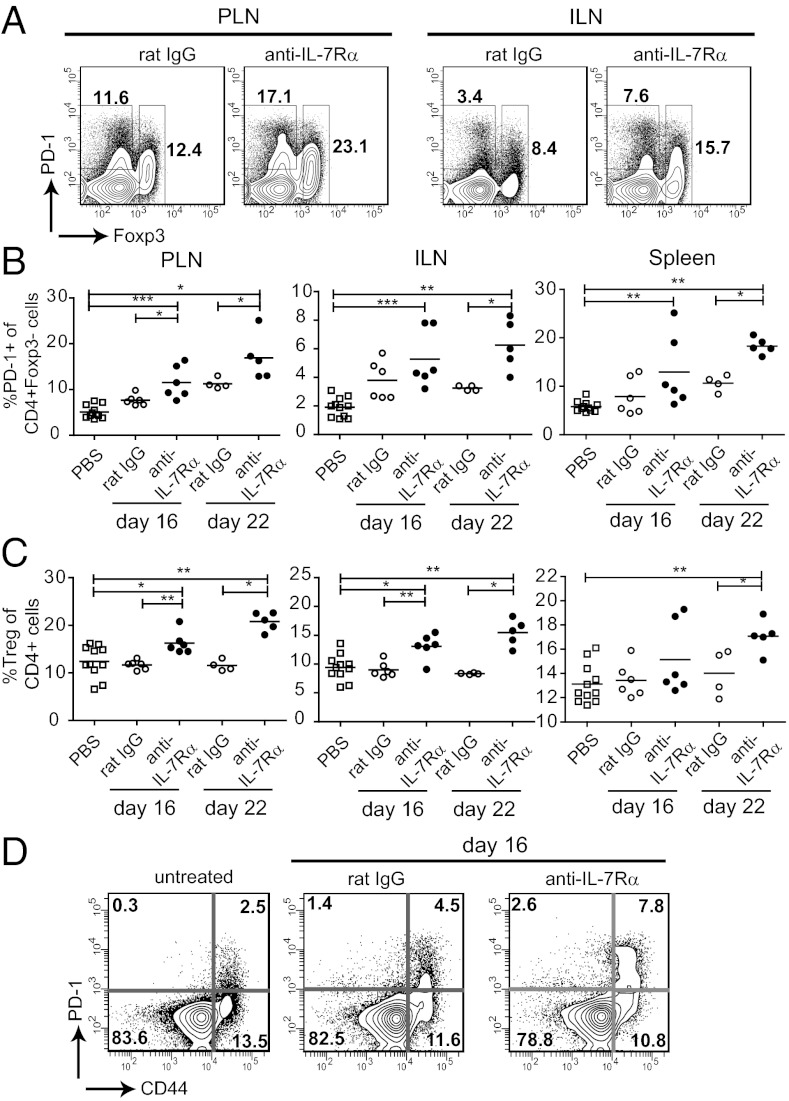
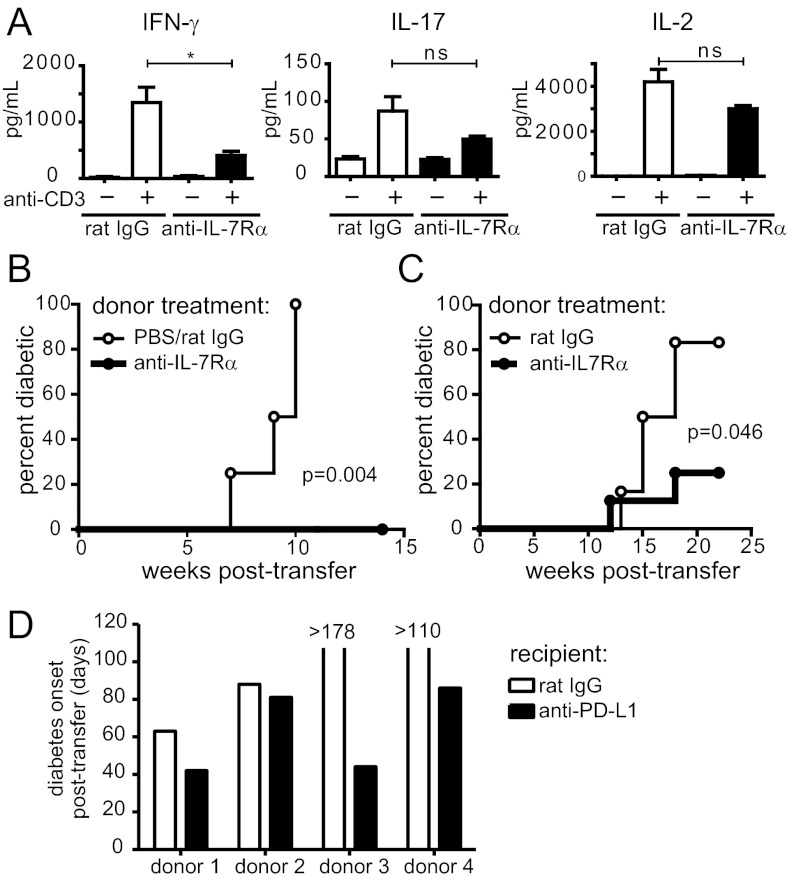
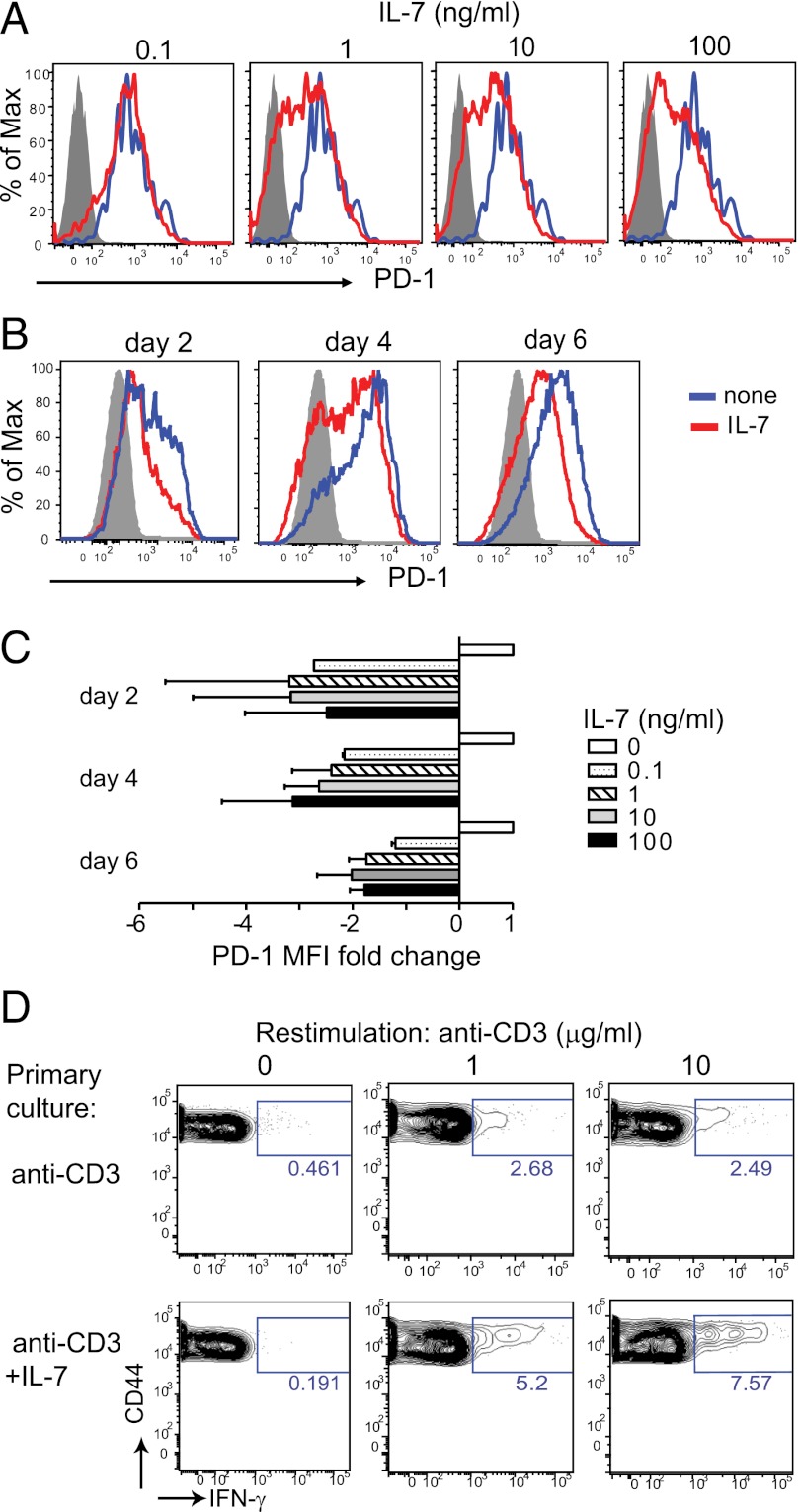
To protect the organism against autoimmunity, self-reactive effector/memory T cells (T(E/M)) are controlled by cell-intrinsic and -extrinsic regulatory mechanisms. However, how some T(E/M) cells escape regulation and cause autoimmune disease is currently not understood. Here we show that blocking IL-7 receptor-α (IL-7Rα) with monoclonal antibodies in nonobese diabetic (NOD) mice prevented autoimmune diabetes and, importantly, reversed disease in new-onset diabetic mice. Surprisingly, IL-7-deprived diabetogenic T(E/M) cells remained present in the treated animals but showed increased expression of the inhibitory receptor Programmed Death 1 (PD-1) and reduced IFN-γ production. Conversely, IL-7 suppressed PD-1 expression on activated T cells in vitro. Adoptive transfer experiments revealed that T(E/M) cells from anti-IL-7Rα-treated mice had lost their pathogenic potential, indicating that absence of IL-7 signals induces cell-intrinsic tolerance. In addition to this mechanism, IL-7Rα blockade altered the balance of regulatory T cells and T(E/M) cells, hence promoting cell-extrinsic regulation and further increasing the threshold for diabetogenic T-cell activation. Our data demonstrate that IL-7 contributes to the pathogenesis of autoimmune diabetes by enabling T(E/M) cells to remain in a functionally competent state and suggest IL-7Rα blockade as a therapy for established T-cell-dependent autoimmune diseases.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
IL-7 receptor α blockade, an off-switch for autoreactive T cells.Proc Natl Acad Sci U S A. 2012 Jul 31;109(31):12270-1. doi: 10.1073/pnas.1209749109. Epub 2012 Jul 23. Proc Natl Acad Sci U S A. 2012. PMID: 22826232 Free PMC article. No abstract available.
-
Autoimmune disease: Targeting IL-7 reverses type 1 diabetes.Nat Rev Drug Discov. 2012 Aug;11(8):599. doi: 10.1038/nrd3805. Nat Rev Drug Discov. 2012. PMID: 22850777 No abstract available.
References
-
- Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29:848–862. - PubMed
-
- Pellegrini M, et al. Adjuvant IL-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies. Nat Med. 2009;15:528–536. - PubMed
-
- Pellegrini M, et al. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell. 2011;144:601–613. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
