

Relocalization of junctional adhesion molecule A during inflammatory stimulation of brain endothelial cells
- PMID: 22733993
- PMCID: PMC3422009
- DOI: 10.1128/MCB.06678-11
Relocalization of junctional adhesion molecule A during inflammatory stimulation of brain endothelial cells
Abstract
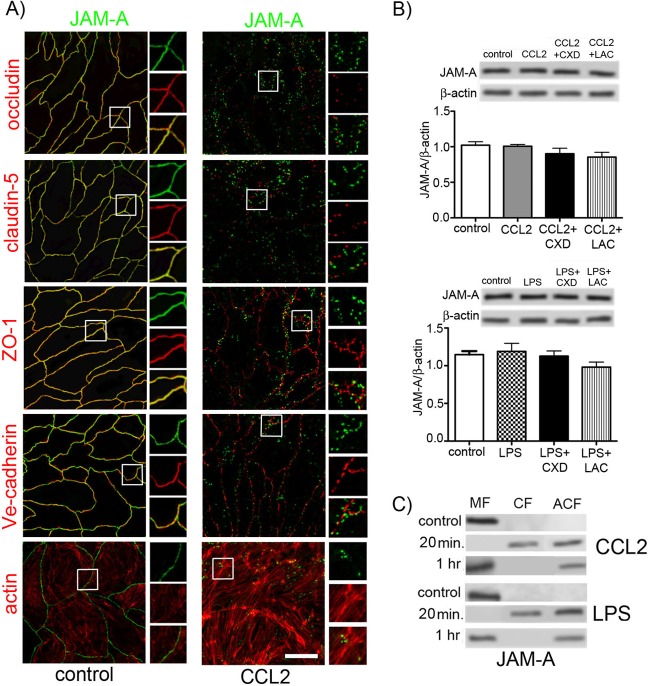
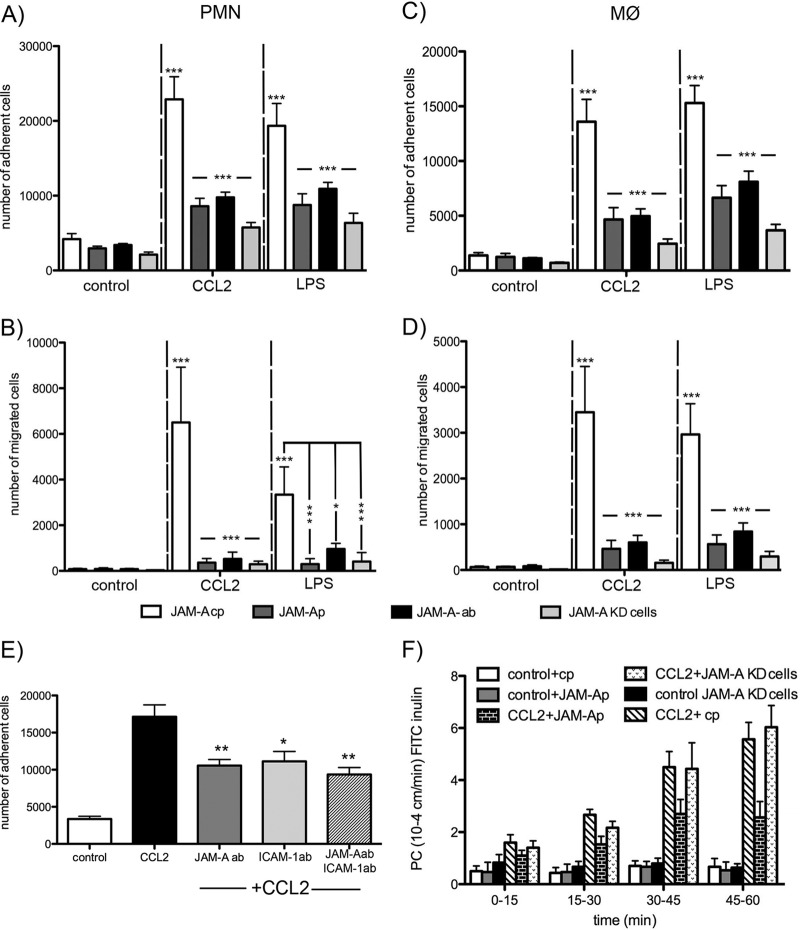
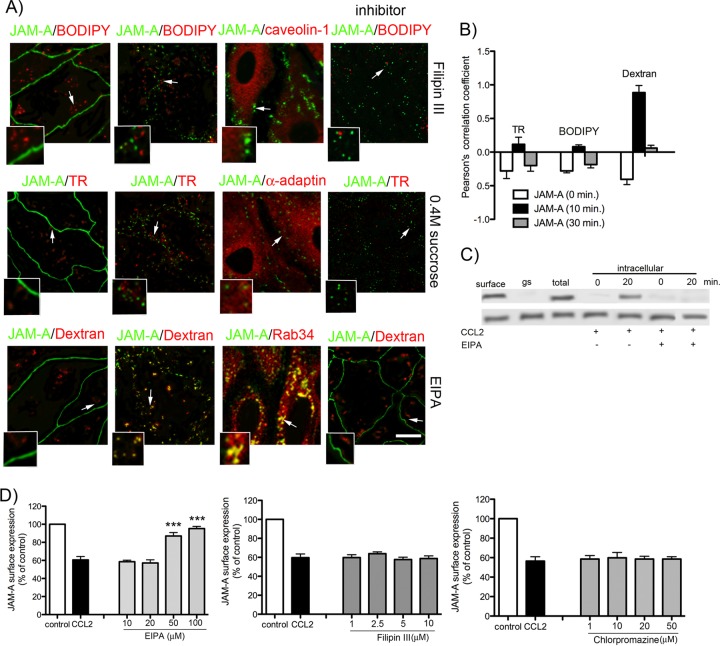
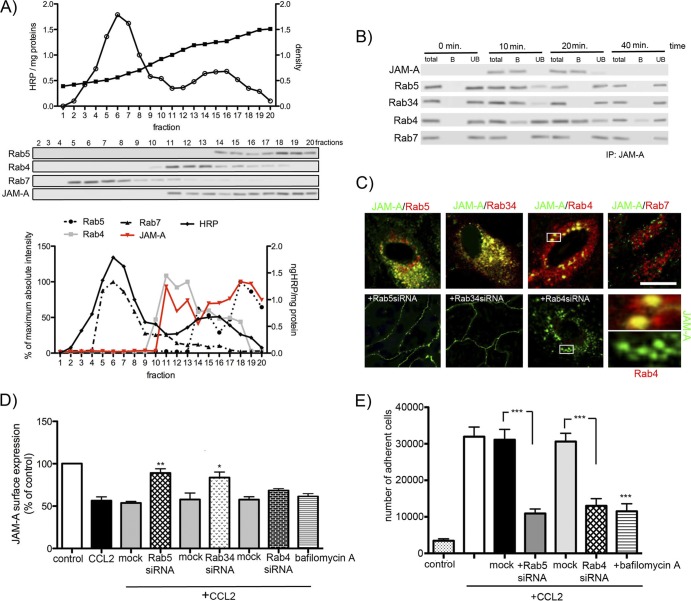
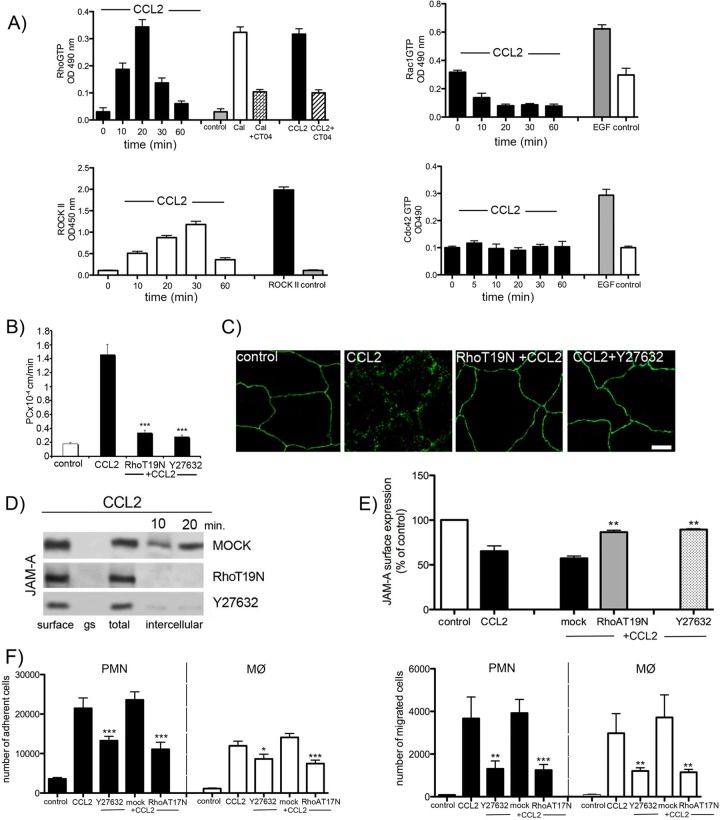
Junctional adhesion molecule A (JAM-A) is a unique tight junction (TJ) transmembrane protein that under basal conditions maintains endothelial cell-cell interactions but under inflammatory conditions acts as a leukocyte adhesion molecule. This study investigates the fate of JAM-A during inflammatory TJ complex remodeling and paracellular route formation in brain endothelial cells. The chemokine (C-C motif) ligand 2 (CCL2) induced JAM-A redistribution from the interendothelial cell area to the apical surface, where JAM-A played a role as a leukocyte adhesion molecule participating in transendothelial cell migration of neutrophils and monocytes. JAM-A redistribution was associated with internalization via macropinocytosis during paracellular route opening. A tracer study with dextran-Texas Red indicated that internalization occurred within a short time period (~10 min) by dextran-positive vesicles and then became sorted to dextran-positive/Rab34-positive/Rab5-positive vesicles and then Rab4-positive endosomes. By ~20 min, most internalized JAM-A moved to the brain endothelial cell apical membrane. Treatment with a macropinocytosis inhibitor, 5-(N-ethyl-N-isopropyl)amiloride, or Rab5/Rab4 depletion with small interfering RNA oligonucleotides prevented JAM-A relocalization, suggesting that macropinocytosis and recycling to the membrane surface occur during JAM-A redistribution. Analysis of the signaling pathways indicated involvement of RhoA and Rho kinase in JAM-A relocalization. These data provide new insights into the molecular and cellular mechanisms involved in blood-brain barrier remodeling during inflammation.
Figures


References
-
- Babinska A, et al. 2007. The F11 receptor (F11R/JAM-A) in atherothrombosis: overexpression of F11R in atherosclerotic plaques. Thromb. Haemost. 97: 272– 281 - PubMed
-
- Bazzoni G. 2011. Pathobiology of junctional adhesion molecules. Antioxid. Redox Signal. 15: 1221– 1234 - PubMed
-
- Bazzoni G, Dejana E. 2004. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol. Rev. 84: 869– 901 - PubMed
-
- Bazzoni G, et al. 2000. Homophilic interaction of junctional adhesion molecule. J. Biol. Chem. 275: 30970– 30976 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases