

G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization
- PMID: 22736029
- PMCID: PMC3441120
- DOI: 10.1093/hmg/dds244
G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization
Abstract
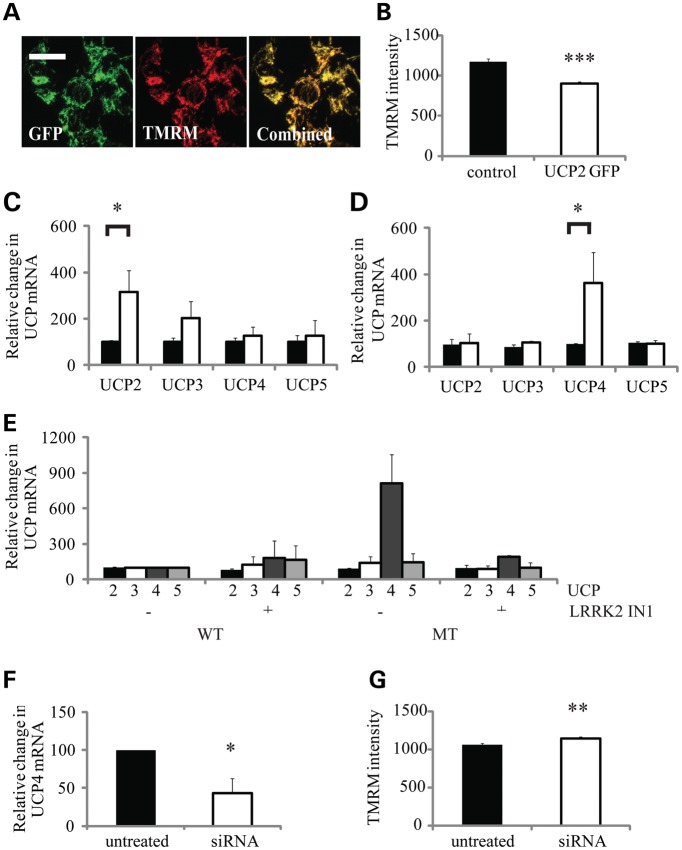
The G2019S leucine rich repeat kinase 2 (LRRK2) mutation is the most common genetic cause of Parkinson's disease (PD), clinically and pathologically indistinguishable from idiopathic PD. Mitochondrial abnormalities are a common feature in PD pathogenesis and we have investigated the impact of G2019S mutant LRRK2 expression on mitochondrial bioenergetics. LRRK2 protein expression was detected in fibroblasts and lymphoblasts at levels higher than those observed in the mouse brain. The presence of G2019S LRRK2 mutation did not influence LRRK2 expression in fibroblasts. However, the expression of the G2019S LRRK2 mutation in both fibroblast and neuroblastoma cells was associated with mitochondrial uncoupling. This was characterized by decreased mitochondrial membrane potential and increased oxygen utilization under basal and oligomycin-inhibited conditions. This resulted in a decrease in cellular ATP levels consistent with compromised cellular function. This uncoupling of mitochondrial oxidative phosphorylation was associated with a cell-specific increase in uncoupling protein (UCP) 2 and 4 expression. Restoration of mitochondrial membrane potential by the UCP inhibitor genipin confirmed the role of UCPs in this mechanism. The G2019S LRRK2-induced mitochondrial uncoupling and UCP4 mRNA up-regulation were LRRK2 kinase-dependent, whereas endogenous LRRK2 levels were required for constitutive UCP expression. We propose that normal mitochondrial function was deregulated by the expression of G2019S LRRK2 in a kinase-dependent mechanism that is a modification of the normal LRRK2 function, and this leads to the vulnerability of selected neuronal populations in PD.
Figures





References
-
- Lees A.J., Hardy J., Revesz T. Parkinson's disease. Lancet. 2009;373:2055–2066. doi:10.1016/S0140-6736(09)60492-X. - DOI - PubMed
-
- Healy D.G., Wood N.W., Schapira A.H. Test for LRRK2 mutations in patients with Parkinson's disease. Pract. Neurol. 2008;8:381–385. doi:10.1136/jnnp.2008.162420. - DOI - PubMed
-
- Marin I. The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol. Biol. Evol. 2006;23:2423–2433. doi:10.1093/molbev/msl114. - DOI - PubMed
-
- Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M.P., Beilina A., Blackinton J., Thomas K.J., et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006;23:329–341. doi:10.1016/j.nbd.2006.04.001. - DOI - PubMed
-
- Ito G., Okai T., Fujino G., Takeda K., Ichijo H., Katada T., Iwatsubo T. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. 2007;46:1380–1388. doi:10.1021/bi061960m. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
