

Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia
- PMID: 22737091
- PMCID: PMC3380828
- DOI: 10.1371/journal.pgen.1002781
Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia
Abstract
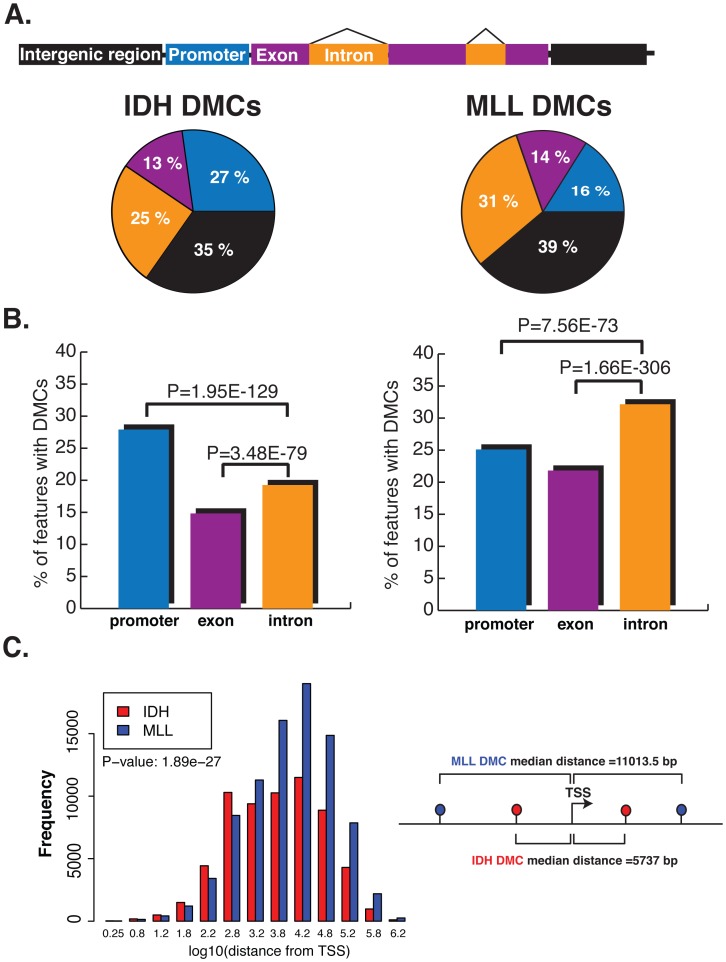
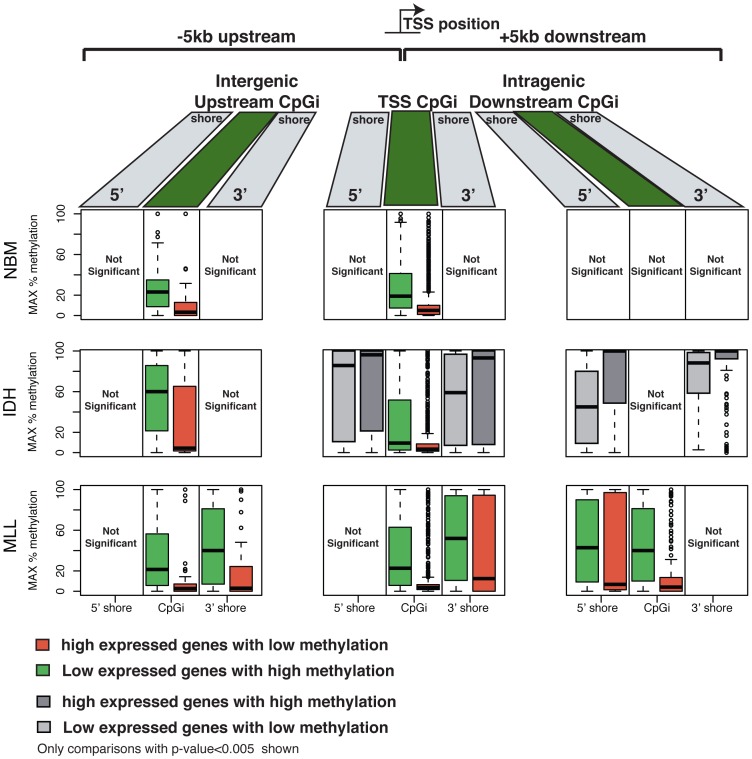
We have developed an enhanced form of reduced representation bisulfite sequencing with extended genomic coverage, which resulted in greater capture of DNA methylation information of regions lying outside of traditional CpG islands. Applying this method to primary human bone marrow specimens from patients with Acute Myelogeneous Leukemia (AML), we demonstrated that genetically distinct AML subtypes display diametrically opposed DNA methylation patterns. As compared to normal controls, we observed widespread hypermethylation in IDH mutant AMLs, preferentially targeting promoter regions and CpG islands neighboring the transcription start sites of genes. In contrast, AMLs harboring translocations affecting the MLL gene displayed extensive loss of methylation of an almost mutually exclusive set of CpGs, which instead affected introns and distal intergenic CpG islands and shores. When analyzed in conjunction with gene expression profiles, it became apparent that these specific patterns of DNA methylation result in differing roles in gene expression regulation. However, despite this subtype-specific DNA methylation patterning, a much smaller set of CpG sites are consistently affected in both AML subtypes. Most CpG sites in this common core of aberrantly methylated CpGs were hypermethylated in both AML subtypes. Therefore, aberrant DNA methylation patterns in AML do not occur in a stereotypical manner but rather are highly specific and associated with specific driving genetic lesions.
Conflict of interest statement
GPS, IK, LZ, and JB are currently or have until recently been employed by Illumina.
Figures
References
-
- Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. - PubMed
-
- Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nature Genetics. 2002;30:41–47. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
