

αSynuclein and Mitochondrial Dysfunction: A Pathogenic Partnership in Parkinson's Disease?
- PMID: 22737587
- PMCID: PMC3377350
- DOI: 10.1155/2012/829207
αSynuclein and Mitochondrial Dysfunction: A Pathogenic Partnership in Parkinson's Disease?
Abstract
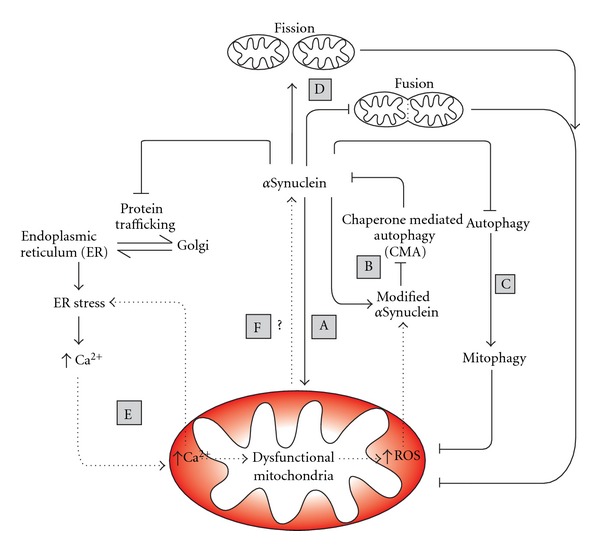
Parkinson's Disease (PD) is a complex, chronic, progressive, and debilitating neurodegenerative disorder. Neither a cure nor effective long-term therapy exist and the lack of knowledge of the molecular mechanisms responsible for PD development is a major impediment to therapeutic advances. The protein αSynuclein is a central component in PD pathogenesis yet its cellular targets and mechanism of toxicity remains unknown. Mitochondrial dysfunction is also a common theme in PD patients and this review explores the strong possibility that αSynuclein and mitochondrial dysfunction have an inter-relationship responsible for underlying the disease pathology. Amplifying cycles of mitochondrial dysfunction and αSynuclein toxicity can be envisaged, with either being the disease-initiating factor yet acting together during disease progression. Multiple potential mechanisms exist in which mitochondrial dysfunction and αSynuclein could interact to exacerbate their neurodegenerative properties. Candidates discussed within this review include autophagy, mitophagy, mitochondrial dynamics/fusion/fission, oxidative stress and reactive oxygen species, endoplasmic reticulum stress, calcium, nitrosative stress and αSynuclein Oligomerization.
Figures
Similar articles
-
Dynamin-related protein 1: A protein critical for mitochondrial fission, mitophagy, and neuronal death in Parkinson's disease.Pharmacol Res. 2020 Jan;151:104553. doi: 10.1016/j.phrs.2019.104553. Epub 2019 Nov 21. Pharmacol Res. 2020. PMID: 31760107 Review.
-
High expression levels of the D686N Parkinson's disease mutation in VPS35 induces α-synuclein-dependent toxicity in yeast.Mol Med Rep. 2017 Jul;16(1):254-262. doi: 10.3892/mmr.2017.6551. Epub 2017 May 9. Mol Med Rep. 2017. PMID: 28487947 Free PMC article.
-
Current perspective of mitochondrial biology in Parkinson's disease.Neurochem Int. 2018 Jul;117:91-113. doi: 10.1016/j.neuint.2018.03.001. Epub 2018 Mar 14. Neurochem Int. 2018. PMID: 29550604 Free PMC article. Review.
-
Mitochondria: A Therapeutic Target for Parkinson's Disease?Int J Mol Sci. 2015 Sep 1;16(9):20704-30. doi: 10.3390/ijms160920704. Int J Mol Sci. 2015. PMID: 26340618 Free PMC article. Review.
-
Mitochondrial biogenesis: pharmacological approaches.Curr Pharm Des. 2014;20(35):5507-9. doi: 10.2174/138161282035140911142118. Curr Pharm Des. 2014. PMID: 24606795
Cited by
-
Inhibition of neuroinflammation and mitochondrial dysfunctions by carbenoxolone in the rotenone model of Parkinson's disease.Mol Neurobiol. 2015 Feb;51(1):209-19. doi: 10.1007/s12035-014-8769-7. Epub 2014 Jun 20. Mol Neurobiol. 2015. PMID: 24946750
-
Individual Amino Acid Supplementation Can Improve Energy Metabolism and Decrease ROS Production in Neuronal Cells Overexpressing Alpha-Synuclein.Neuromolecular Med. 2017 Sep;19(2-3):322-344. doi: 10.1007/s12017-017-8448-8. Epub 2017 Jun 15. Neuromolecular Med. 2017. PMID: 28620826
-
Coenzyme Q10 depletion in medical and neuropsychiatric disorders: potential repercussions and therapeutic implications.Mol Neurobiol. 2013 Dec;48(3):883-903. doi: 10.1007/s12035-013-8477-8. Epub 2013 Jun 13. Mol Neurobiol. 2013. PMID: 23761046 Review.
-
Mortal engines: Mitochondrial bioenergetics and dysfunction in neurodegenerative diseases.Pharmacol Res. 2018 Dec;138:2-15. doi: 10.1016/j.phrs.2018.08.010. Epub 2018 Aug 23. Pharmacol Res. 2018. PMID: 30144530 Free PMC article. Review.
-
Selective brain penetrable Nurr1 transactivator for treating Parkinson's disease.Oncotarget. 2016 Feb 16;7(7):7469-79. doi: 10.18632/oncotarget.7191. Oncotarget. 2016. PMID: 26862735 Free PMC article.
References
-
- Thomas B, Beal MF. Parkinson’s disease. Human Molecular Genetics. 2007;16:R183–R194. - PubMed
-
- Schapira AHV. Mitochondrial dysfunction in Parkinson’s disease. Cell Death and Differentiation. 2007;14(7):1261–1266. - PubMed
-
- Mizuno Y, Ohta S, Tanaka M, et al. Deficiencies in Complex I subunits of the respiratory chain in Parkinson's disease. Biochemical and Biophysical Research Communications. 1989;163:1450–1455. - PubMed
-
- Schapira AHV, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial Complex I deficiency in Parkinson’s disease. The Lancet. 1989;1(8649):p. 1269. - PubMed
-
- Schapira AHV, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial Complex I deficiency in Parkinson’s disease. Journal of Neurochemistry. 1990;54(3):823–827. - PubMed
LinkOut - more resources
Full Text Sources
