

Targeting eNOS in pancreatic cancer
- PMID: 22738914
- PMCID: PMC3749841
- DOI: 10.1158/0008-5472.CAN-12-0057
Targeting eNOS in pancreatic cancer
Abstract
Mortality from pancreatic ductal adenocarcinoma cancer (PDAC) is among the highest of any cancer and frontline therapy has changed little in years. Activation of endothelial nitric oxide synthase (eNOS, NOS3, or NOS III) has been implicated recently in the pathogenesis of PDACs. In this study, we used genetically engineered mouse and human xenograft models to evaluate the consequences of targeting eNOS in PDACs. Genetic deficiency in eNOS limited the development of preinvasive pancreatic lesions and trended toward an extended lifespan in mice with advanced pancreatic cancer. These effects were also observed upon oral administration of the clinically evaluated NOS small molecule inhibitor N(G)-nitro-L-arginine methyl ester (l-NAME). Similarly, other transgenic models of oncogenic KRas-driven tumors responded to l-NAME treatment. Finally, these results were recapitulated in xenograft models of human pancreatic cancer, in which l-NAME was found to broadly inhibit tumorigenic growth. Taken together, our findings offer preclinical proof-of-principle to repurpose l-NAME for clinical investigations in treatment of PDACs and possibly other KRas-driven human cancers.
©2012 AACR.
Conflict of interest statement
Figures
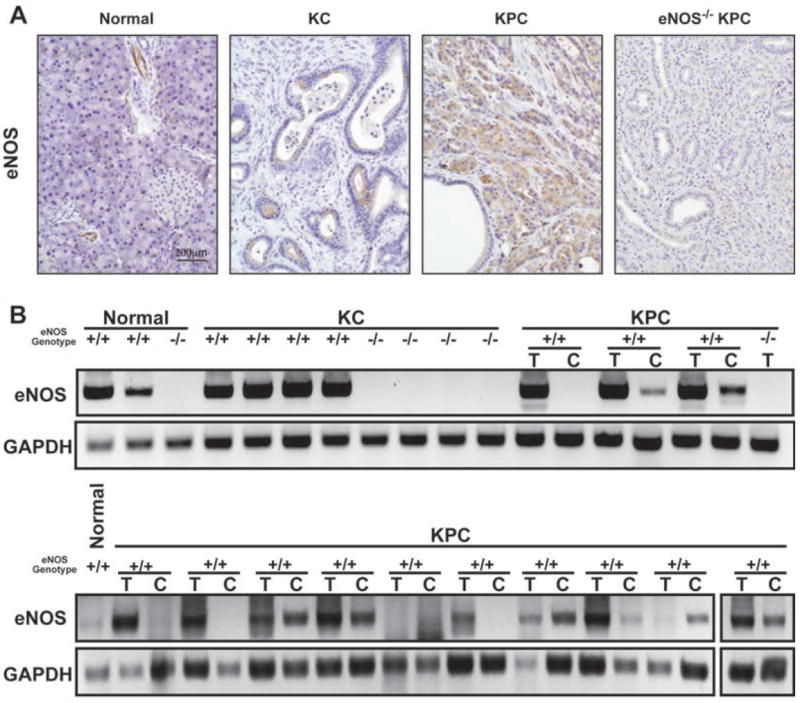
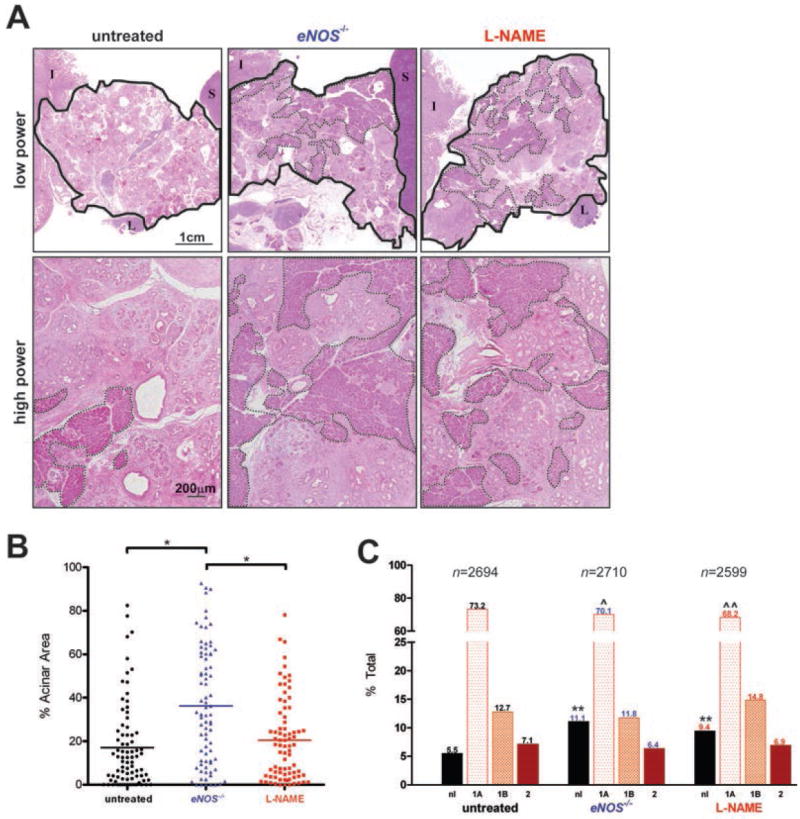
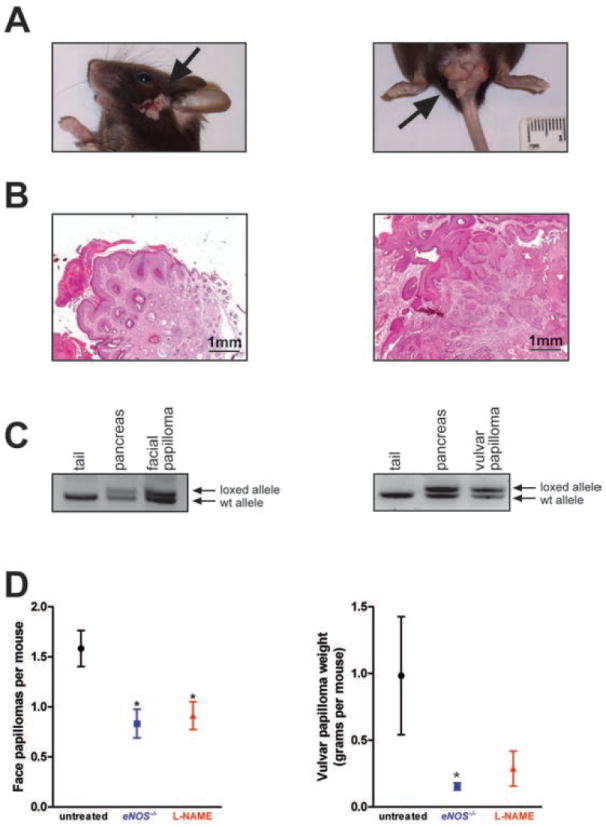
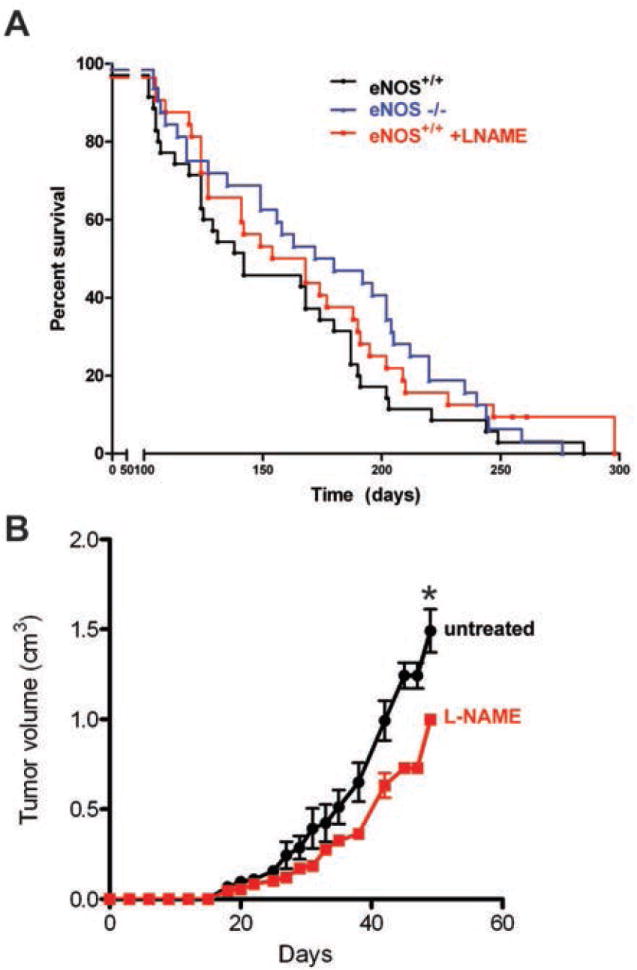
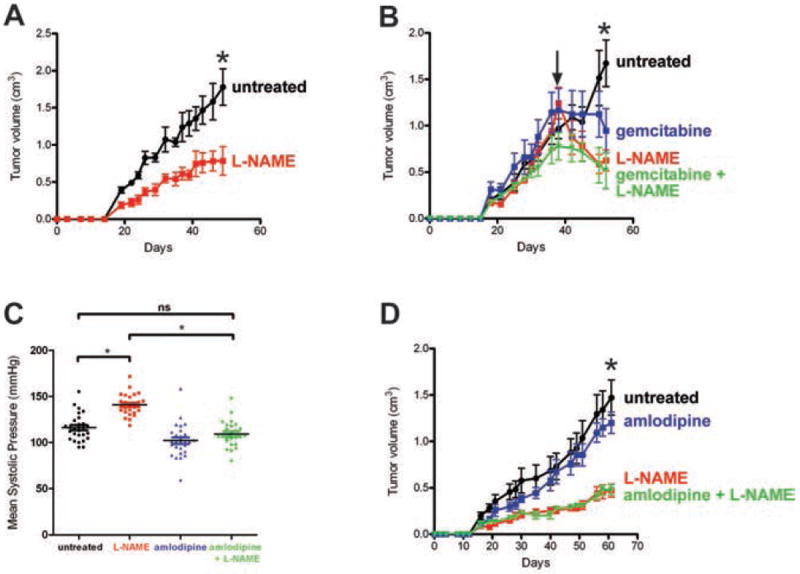
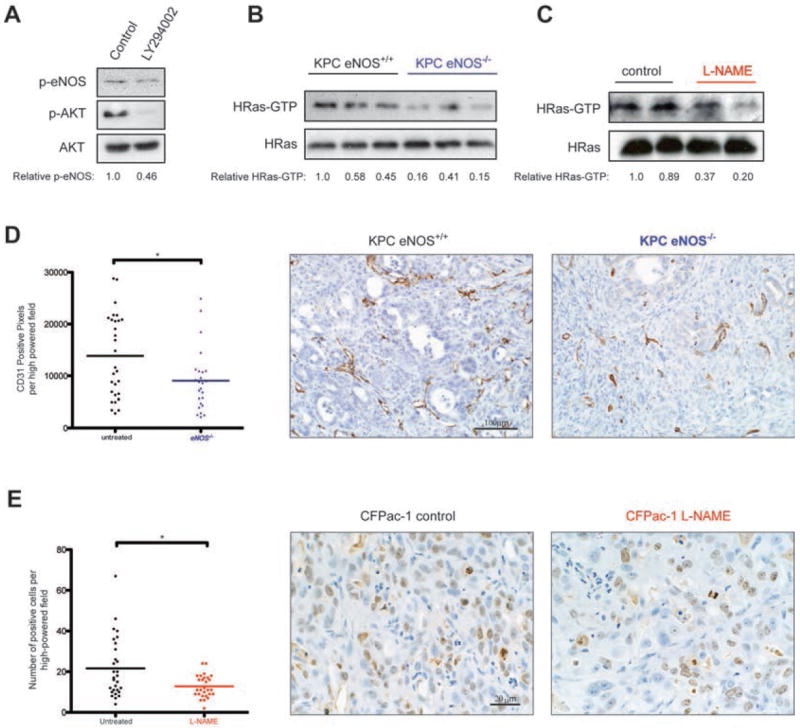
References
-
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. - PubMed
-
- Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 364:1817–25. - PubMed
-
- Ko AH, Tempero MA. Treatment of metastatic pancreatic cancer. J Natl Compr Canc Netw. 2005;3:627–36. - PubMed
-
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. - PubMed
-
- Qian J, Niu J, Li M, Chiao PJ, Tsao MS. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 2005;65:5045–53. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
