

14q12 and severe Rett-like phenotypes: new clinical insights and physical mapping of FOXG1-regulatory elements
- PMID: 22739344
- PMCID: PMC3499785
- DOI: 10.1038/ejhg.2012.127
14q12 and severe Rett-like phenotypes: new clinical insights and physical mapping of FOXG1-regulatory elements
Abstract
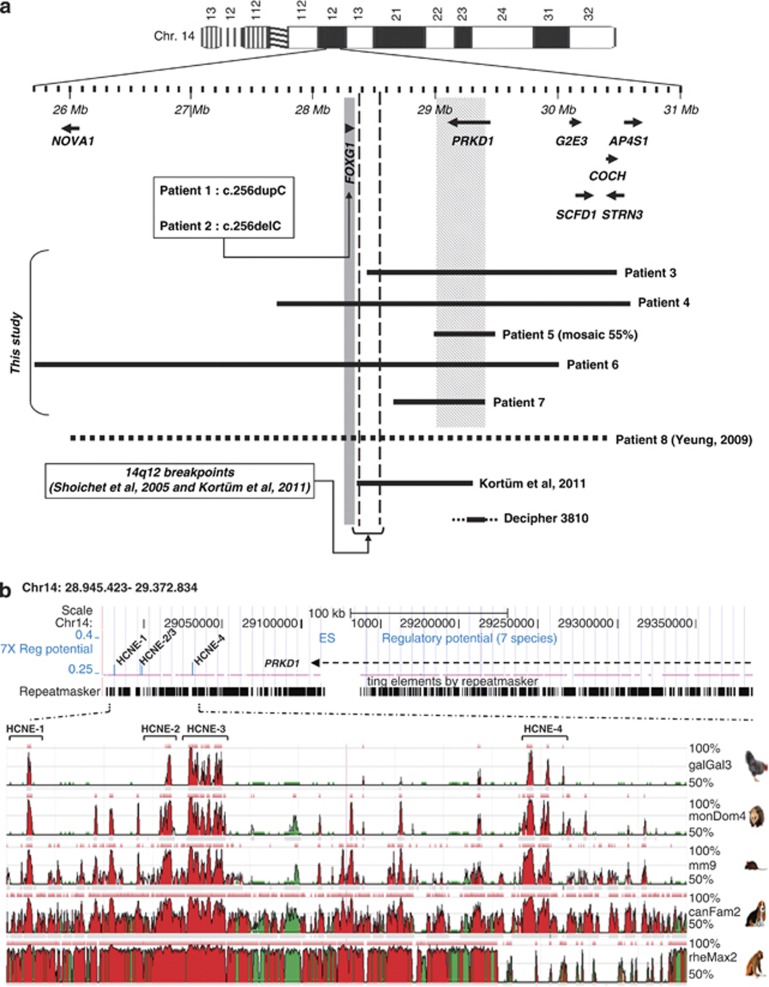
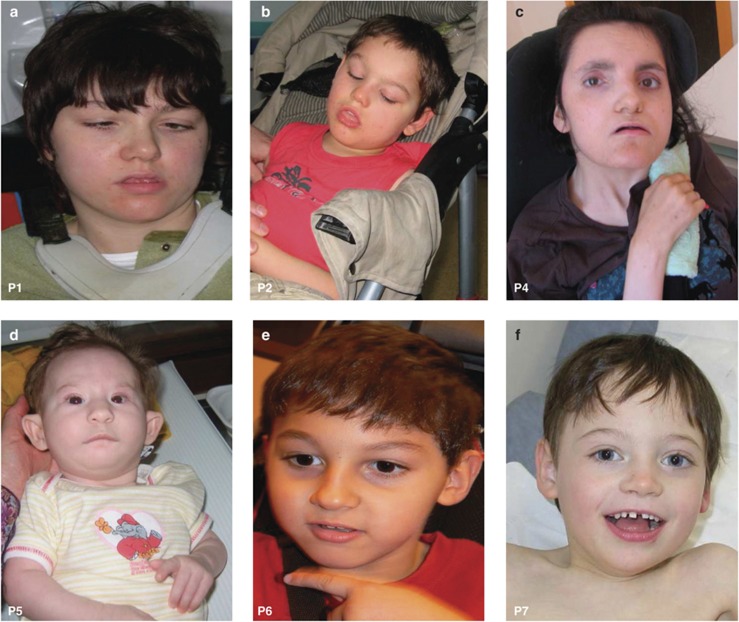
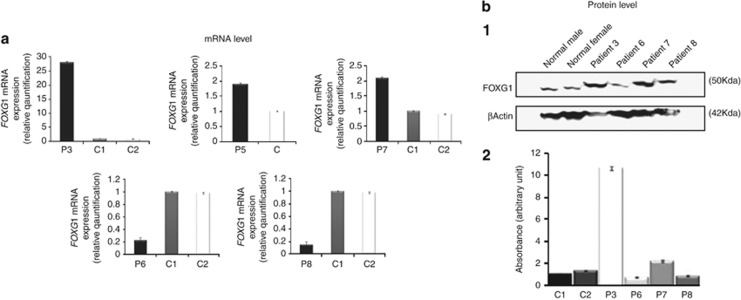
The Forkhead box G1 (FOXG1) gene has been implicated in severe Rett-like phenotypes. It encodes the Forkhead box protein G1, a winged-helix transcriptional repressor critical for forebrain development. Recently, the core FOXG1 syndrome was defined as postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and dysgenesis of the corpus callosum. We present seven additional patients with a severe Rett-like neurodevelopment disorder associated with de novo FOXG1 point mutations (two cases) or 14q12 deletions (five cases). We expand the mutational spectrum in patients with FOXG1-related encephalopathies and precise the core FOXG1 syndrome phenotype. Dysgenesis of the corpus callosum and dyskinesia are not always present in FOXG1-mutated patients. We believe that the FOXG1 gene should be considered in severely mentally retarded patients (no speech-language) with severe acquired microcephaly (-4 to-6 SD) and few clinical features suggestive of Rett syndrome. Interestingly enough, three 14q12 deletions that do not include the FOXG1 gene are associated with phenotypes very reminiscent to that of FOXG1-mutation-positive patients. We physically mapped a putative long-range FOXG1-regulatory element in a 0.43 Mb DNA segment encompassing the PRKD1 locus. In fibroblast cells, a cis-acting regulatory sequence located more than 0.6 Mb away from FOXG1 acts as a silencer at the transcriptional level. These data are important for clinicians and for molecular biologists involved in the management of patients with severe encephalopathies compatible with a FOXG1-related phenotype.
Figures
References
-
- Le Guen T, Fichou Y, Nectoux J, et al. A missense mutation within the fork-head domain of the forkhead box G1 gene (FOXG1) affects its nuclear localization. Hum Mutat. 2011;32:E2026–E2035. - PubMed
-
- Philippe C, Amsallem D, Francannet C, et al. Phenotypic variability in Rett syndrome associated with FOXG1 mutations in females. J Med Genet. 2010;47:59–65. - PubMed
-
- Mencarelli MA, Spanhol-Rosseto A, Artuso R, et al. Novel FOXG1 mutations associated with the congenital variant of Rett syndrome. J Med Genet. 2010;47:49–53. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
