

HtrA2 deficiency causes mitochondrial uncoupling through the F₁F₀-ATP synthase and consequent ATP depletion
- PMID: 22739987
- PMCID: PMC3388244
- DOI: 10.1038/cddis.2012.77
HtrA2 deficiency causes mitochondrial uncoupling through the F₁F₀-ATP synthase and consequent ATP depletion
Abstract
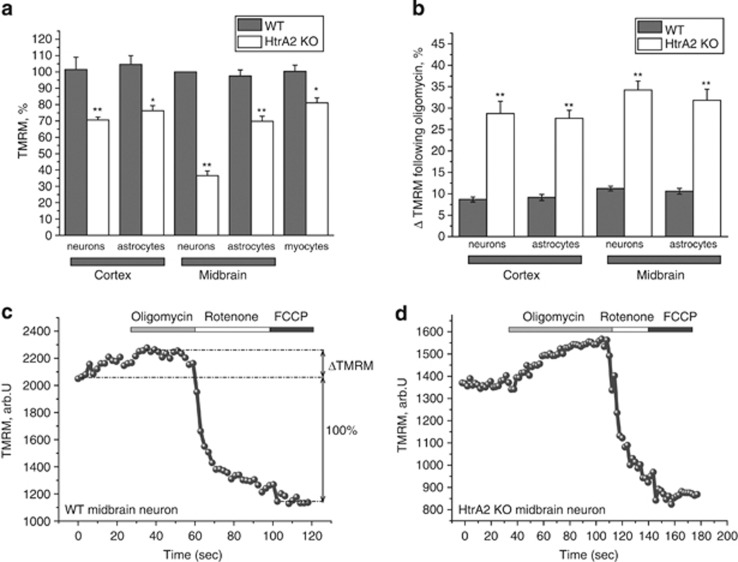
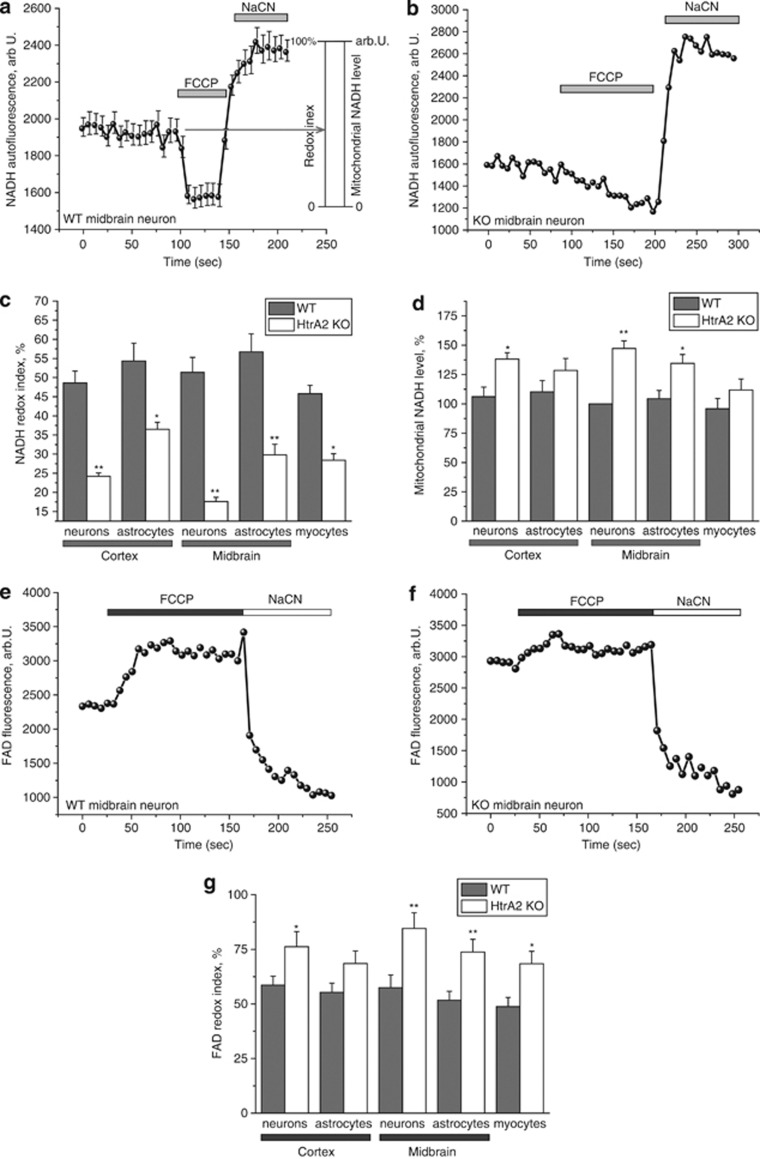
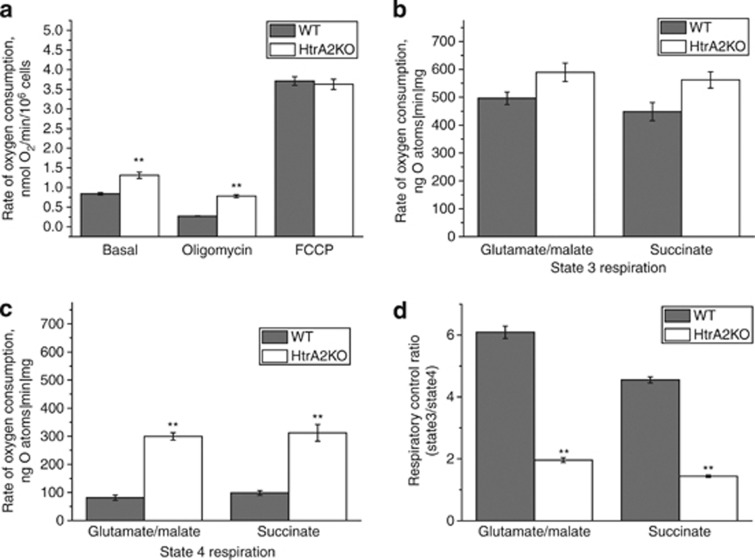
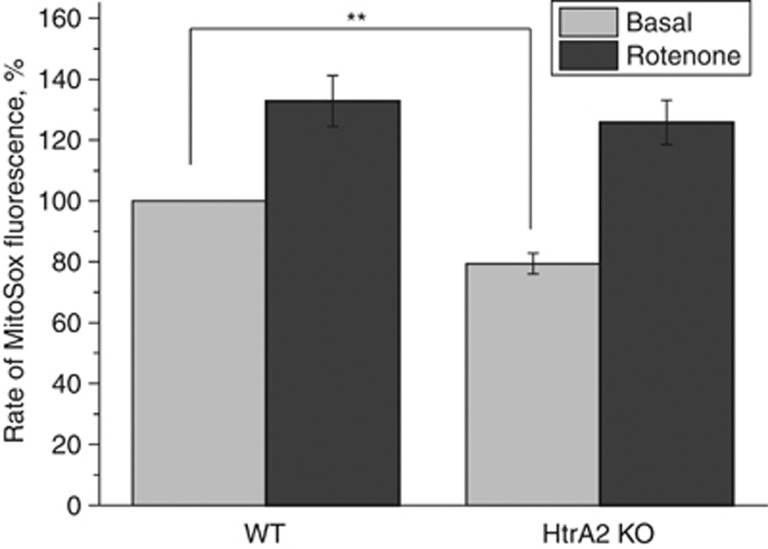
Loss of the mitochondrial protease HtrA2 (Omi) in mice leads to mitochondrial dysfunction, neurodegeneration and premature death, but the mechanism underlying this pathology remains unclear. Using primary cultures from wild-type and HtrA2-knockout mice, we find that HtrA2 deficiency significantly reduces mitochondrial membrane potential in a range of cell types. This depolarisation was found to result from mitochondrial uncoupling, as mitochondrial respiration was increased in HtrA2-deficient cells and respiratory control ratio was dramatically reduced. HtrA2-knockout cells exhibit increased proton translocation through the ATP synthase, in combination with decreased ATP production and truncation of the F1 α-subunit, suggesting the ATP synthase as the source of the proton leak. Uncoupling in the HtrA2-deficient mice is accompanied by altered breathing pattern and, on a cellular level, ATP depletion and vulnerability to chemical ischaemia. We propose that this vulnerability may ultimately cause the neurodegeneration observed in these mice.
Figures
References
-
- Vande Walle L, Lamkanfi M, Vandenabeele P. The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ. 2008;15:45s3–460. - PubMed
-
- Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum Mol Genet. 2005;14:2099–2111. - PubMed
-
- Bogaerts V, Nuytemans K, Reumers J, Pals P, Engelborghs S, Pickut B, et al. Genetic variability in the mitochondrial serine protease HTRA2 contributes to risk for Parkinson disease. Hum Mutat. 2008;29:832–840. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
