

Entry of hepatitis B virus into immortalized human primary hepatocytes by clathrin-dependent endocytosis
- PMID: 22740403
- PMCID: PMC3416113
- DOI: 10.1128/JVI.00873-12
Entry of hepatitis B virus into immortalized human primary hepatocytes by clathrin-dependent endocytosis
Abstract
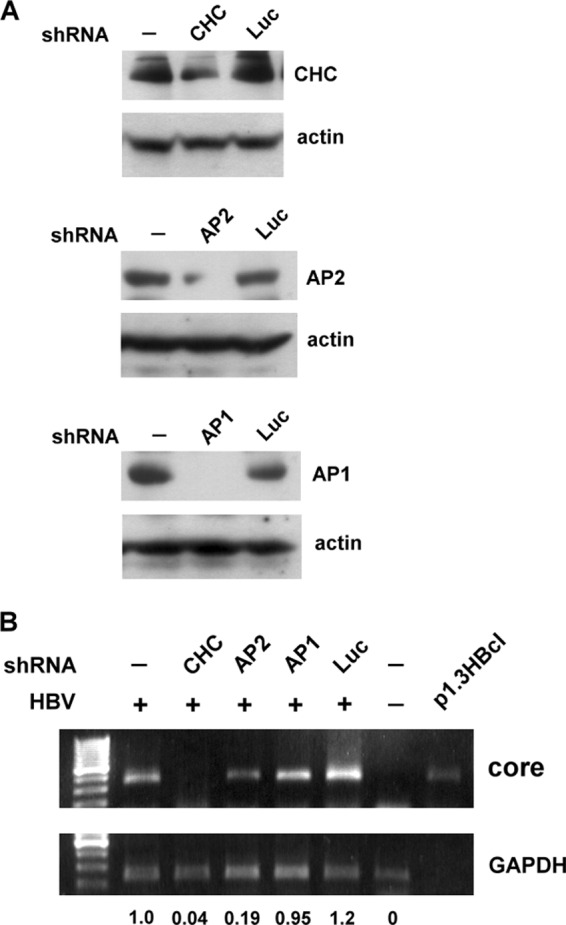
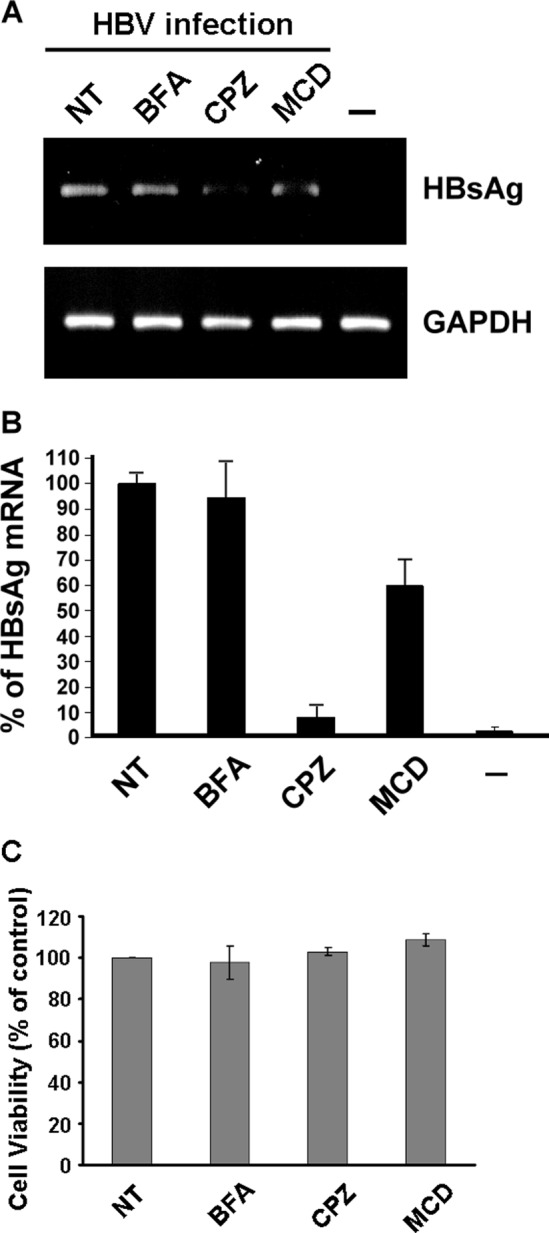
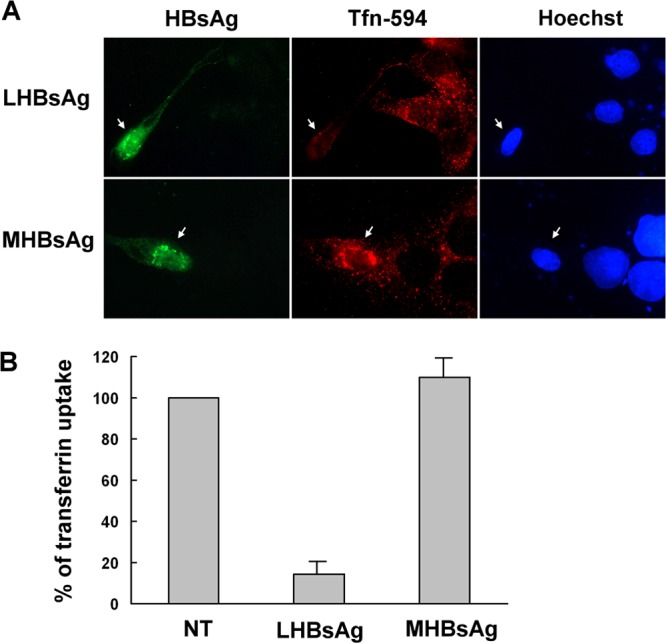
The lack of a suitable in vitro hepatitis B virus (HBV) infectivity model has limited examination of the early stages of the virus-cell interaction. In this study, we used an immortalized cell line derived from human primary hepatocytes, HuS-E/2, to study the mechanism of HBV infection. HBV infection efficiency was markedly increased after dimethyl sulfoxide (DMSO)-induced differentiation of the cells. Transmission electron microscopy demonstrated the presence of intact HBV particles in DMSO-treated HBV-infected HuS-E/2 cells, which could be infected with HBV for up to at least 50 passages. The pre-S1 domain of the large HBsAg (LHBsAg) protein specifically interacted with clathrin heavy chain (CHC) and clathrin adaptor protein AP-2. Short hairpin RNA knockdown of CHC or AP-2 in HuS-E/2 cells significantly reduced their susceptibility to HBV, indicating that both are necessary for HBV infection. Furthermore, HBV entry was inhibited by chlorpromazine, an inhibitor of clathrin-mediated endocytosis. LHBsAg also interfered with the clathrin-mediated endocytosis of transferrin by human hepatocytes. This infection system using an immortalized human primary hepatocyte cell line will facilitate investigations into HBV entry and in devising therapeutic strategies for manipulating HBV-associated liver disorders.
Figures
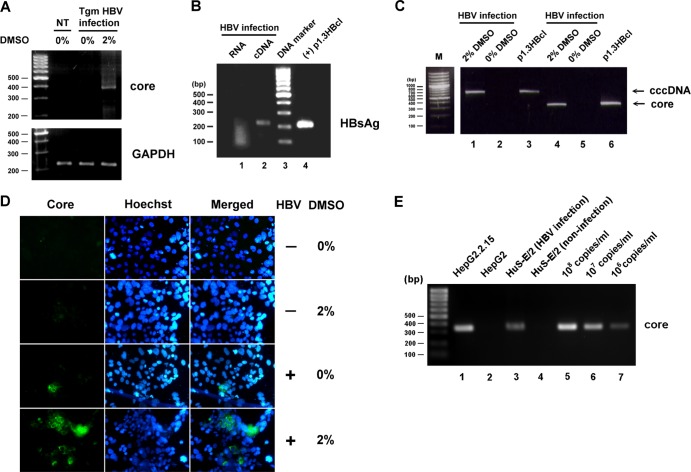
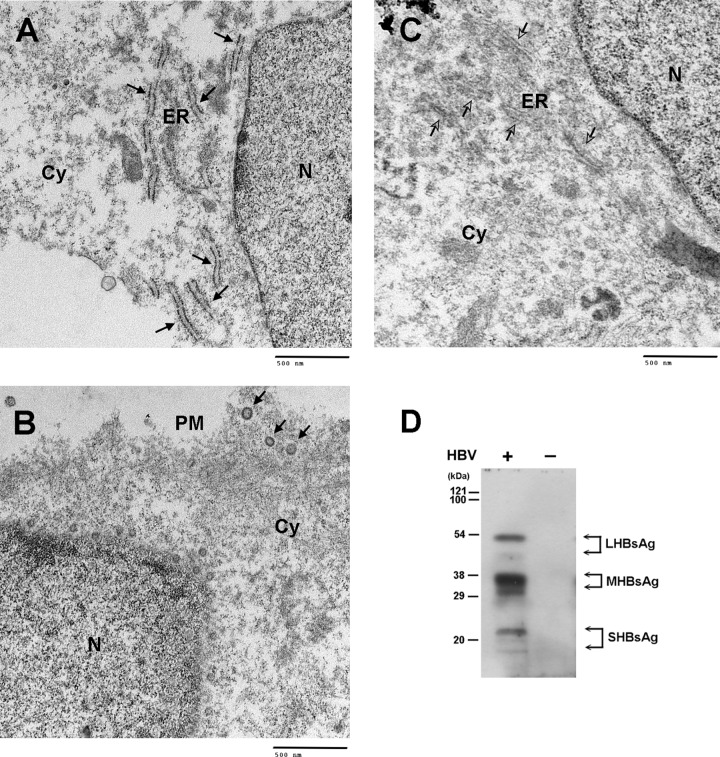
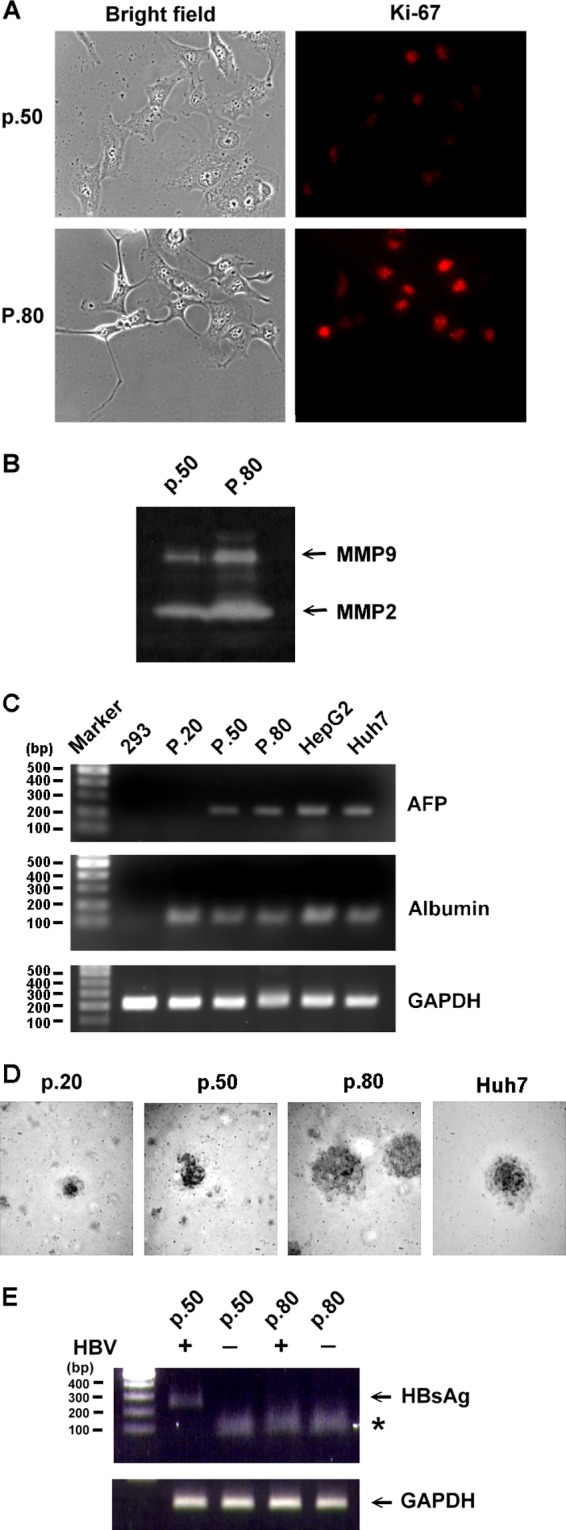
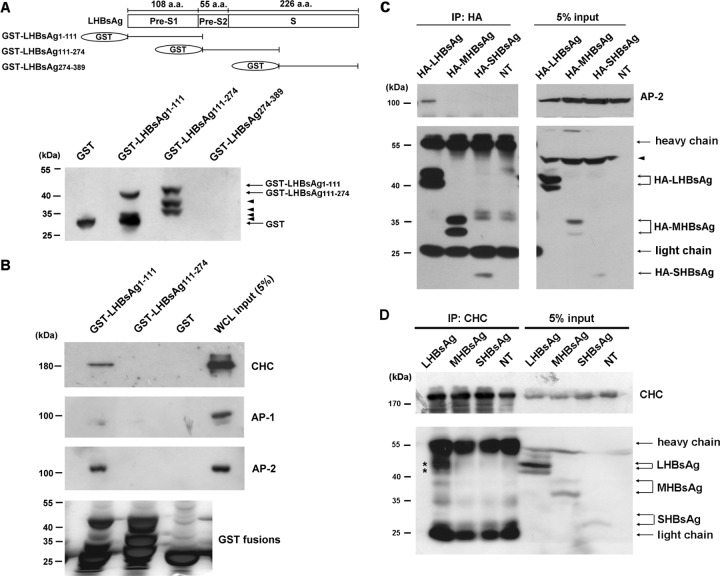
References
-
- Aly HH, et al. 2007. Serum-derived hepatitis C virus infectivity in interferon regulatory factor-7-suppressed human primary hepatocytes. J. Hepatol. 46:26–36 - PubMed
-
- Beasley RP. 1988. Hepatitis B virus. The major etiology of hepatocellular carcinoma. Cancer 61:1942–1956 - PubMed
-
- Bremer CM, Bung C, Kott N, Hardt M, Glebe D. 2009. Hepatitis B virus infection is dependent on cholesterol in the viral envelope. Cell. Microbiol. 11:249–260 - PubMed
-
- Chen CC, et al. 2007. Long-term inhibition of hepatitis B virus in transgenic mice by double-stranded adeno-associated virus 8-delivered short hairpin RNA. Gene Ther. 14:11–19 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
