

Early pathogenesis in the adult-onset neurodegenerative disease amyotrophic lateral sclerosis
- PMID: 22740507
- PMCID: PMC3886915
- DOI: 10.1002/jcb.24234
Early pathogenesis in the adult-onset neurodegenerative disease amyotrophic lateral sclerosis
Abstract
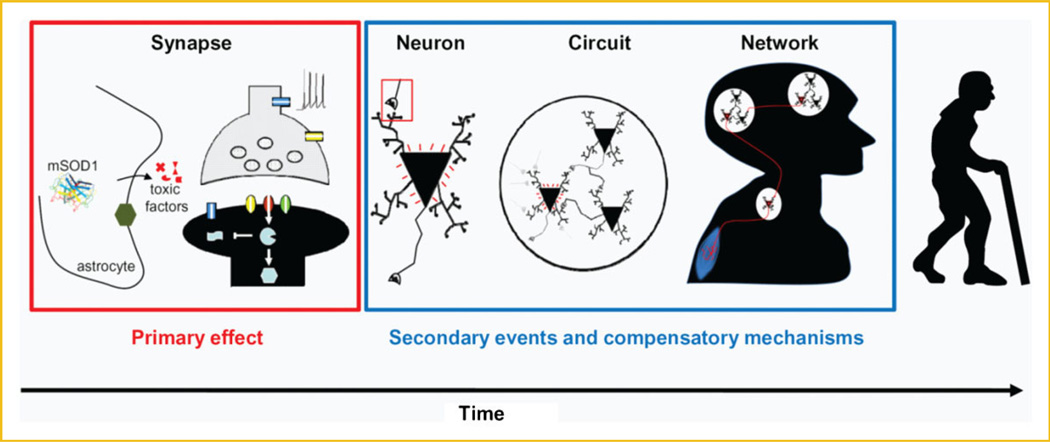
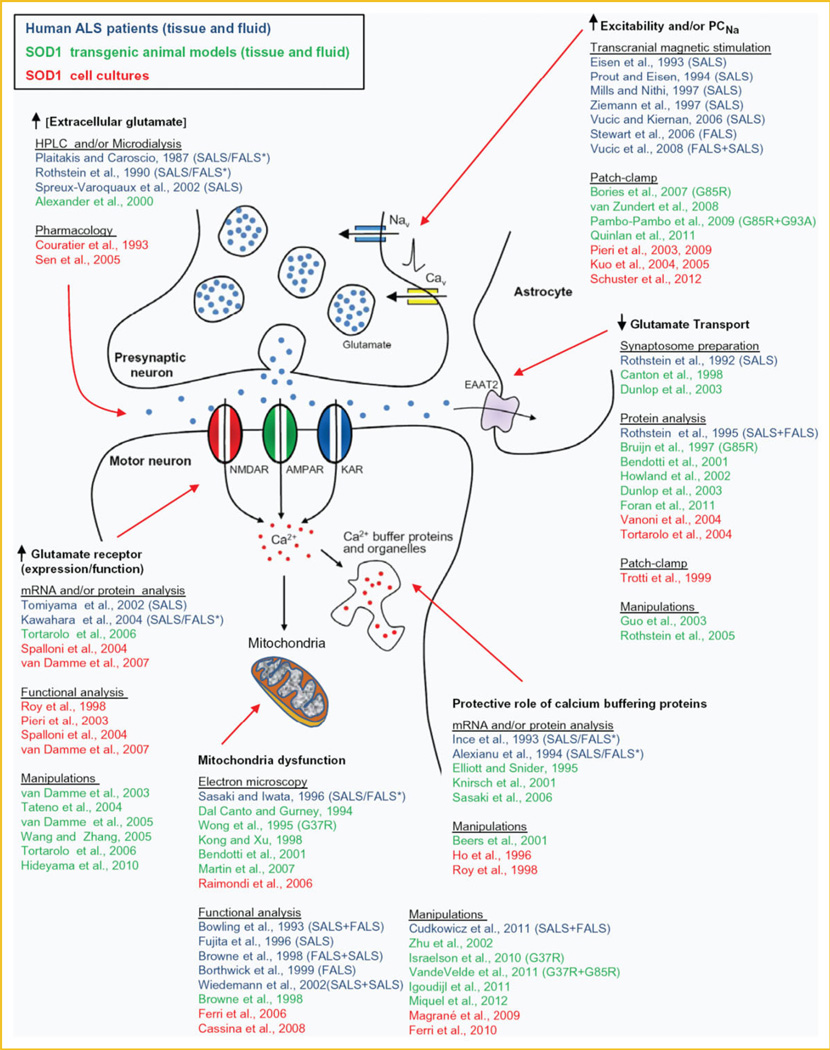
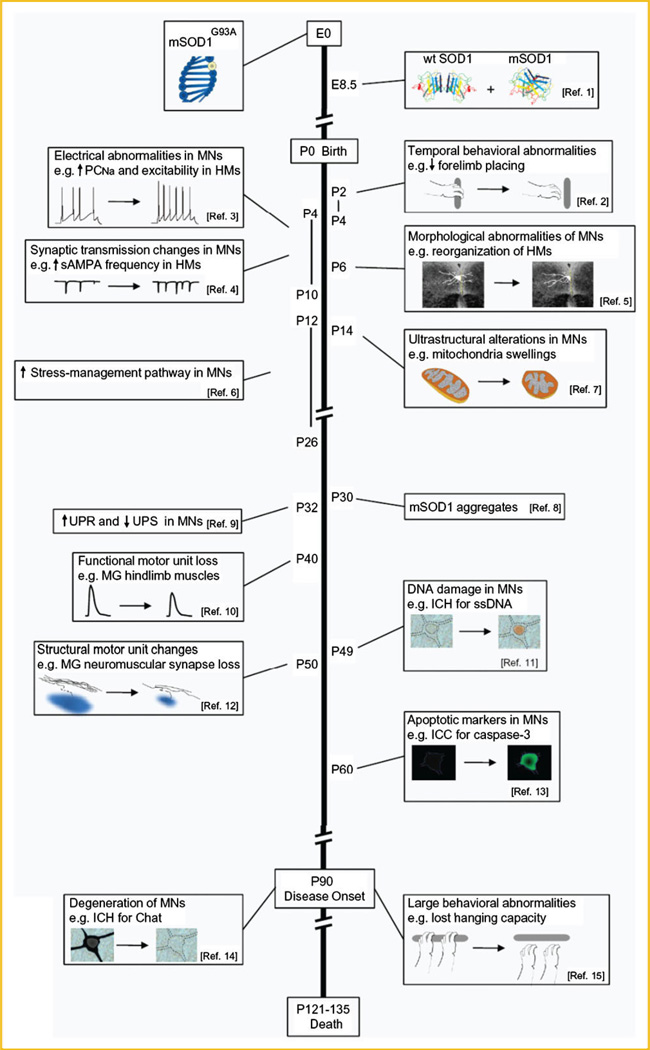
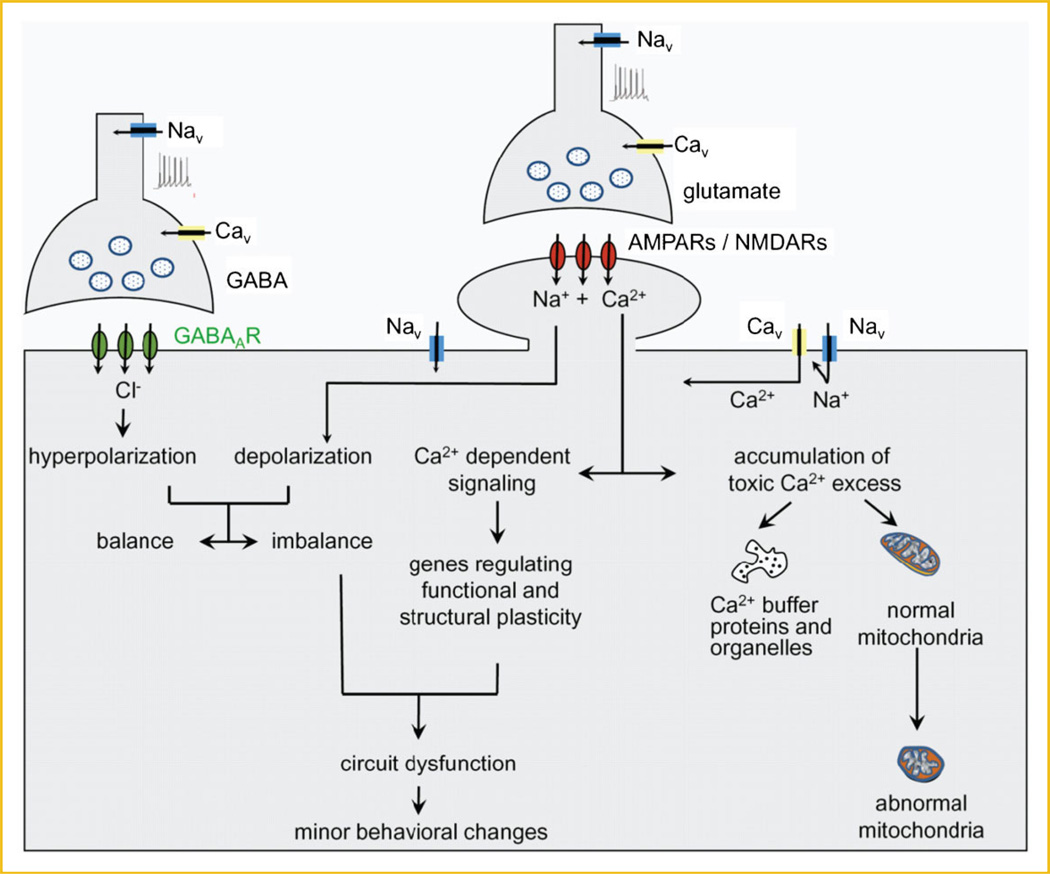
Amyotrophic lateral sclerosis (ALS) is a devastating paralytic disorder caused by dysfunction and degeneration of motor neurons starting in adulthood. Most of our knowledge about the pathophysiological mechanisms of ALS comes from transgenic mice models that emulate a subgroup of familial ALS cases (FALS), with mutations in the gene encoding superoxide dismutase (SOD1). In the more than 15 years since these mice were generated, a large number of abnormal cellular mechanisms underlying motor neuron degeneration have been identified, but to date this effort has led to few improvements in therapy, and no cure. Here, we consider that this surfeit of mechanisms is best interpreted by current insights that suggest a very early initiation of pathology in motor neurons, followed by a diversity of secondary cascades and compensatory mechanisms that mask symptoms for decades, until trauma and/or aging overloads their protective function. This view thus posits that adult-onset ALS is the consequence of processes initiated during early development. In fact, motor neurons in neonatal mutant SOD mice display important alterations in their intrinsic electrical properties, synaptic inputs and morphology that are accompanied by subtle behavioral abnormalities. We consider evidence that human mutant SOD1 protein in neonatal hSOD1(G93A) mice instigates motor neuron degeneration by increasing persistent sodium currents and excitability, in turn altering synaptic circuits that control excessive motor neuron firing and leads to excitotoxicity. We also discuss how therapies that are aimed at suppressing abnormal neuronal activity might effectively mitigate or prevent the onset of irreversible neuronal damage in adulthood. J. Cell. Biochem. 113: 3301-3312, 2012. © 2012 Wiley Periodicals, Inc.
Copyright © 2012 Wiley Periodicals, Inc.
Figures
Similar articles
-
Nuclear localization of human SOD1 and mutant SOD1-specific disruption of survival motor neuron protein complex in transgenic amyotrophic lateral sclerosis mice.J Neuropathol Exp Neurol. 2012 Feb;71(2):162-77. doi: 10.1097/NEN.0b013e318244b635. J Neuropathol Exp Neurol. 2012. PMID: 22249462 Free PMC article.
-
Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1.Neurobiol Dis. 2000 Dec;7(6 Pt B):623-43. doi: 10.1006/nbdi.2000.0299. Neurobiol Dis. 2000. PMID: 11114261
-
Pathological Modification of TDP-43 in Amyotrophic Lateral Sclerosis with SOD1 Mutations.Mol Neurobiol. 2019 Mar;56(3):2007-2021. doi: 10.1007/s12035-018-1218-2. Epub 2018 Jul 7. Mol Neurobiol. 2019. PMID: 29982983 Free PMC article.
-
Early abnormalities in transgenic mouse models of amyotrophic lateral sclerosis.J Physiol Paris. 2006 Mar-May;99(2-3):211-20. doi: 10.1016/j.jphysparis.2005.12.014. Epub 2006 Jan 30. J Physiol Paris. 2006. PMID: 16448809 Review.
-
Rodent Models of Amyotrophic Lateral Sclerosis.Curr Protoc Pharmacol. 2015 Jun 1;69:5.67.1-5.67.21. doi: 10.1002/0471141755.ph0567s69. Curr Protoc Pharmacol. 2015. PMID: 26344214 Free PMC article. Review.
Cited by
-
Emerging mechanisms of molecular pathology in ALS.J Clin Invest. 2015 May;125(5):1767-79. doi: 10.1172/JCI71601. Epub 2015 May 1. J Clin Invest. 2015. PMID: 25932674 Free PMC article. Review.
-
Homeostatic dysregulation in membrane properties of masticatory motoneurons compared with oculomotor neurons in a mouse model for amyotrophic lateral sclerosis.J Neurosci. 2015 Jan 14;35(2):707-20. doi: 10.1523/JNEUROSCI.1682-14.2015. J Neurosci. 2015. PMID: 25589764 Free PMC article.
-
Marked changes in dendritic structure and spine density precede significant neuronal death in vulnerable cortical pyramidal neuron populations in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis.Acta Neuropathol Commun. 2016 Aug 4;4(1):77. doi: 10.1186/s40478-016-0347-y. Acta Neuropathol Commun. 2016. PMID: 27488828 Free PMC article.
-
Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability.J Neurophysiol. 2013 Jun;109(11):2803-14. doi: 10.1152/jn.00500.2012. Epub 2013 Mar 13. J Neurophysiol. 2013. PMID: 23486205 Free PMC article.
-
Excessive release of inorganic polyphosphate by ALS/FTD astrocytes causes non-cell-autonomous toxicity to motoneurons.Neuron. 2022 May 18;110(10):1656-1670.e12. doi: 10.1016/j.neuron.2022.02.010. Epub 2022 Mar 10. Neuron. 2022. PMID: 35276083 Free PMC article.
References
-
- Amendola J, Durand J. Morphological differences between wild-type and transgenic superoxide dismutase 1 lumbar motoneurons in postnatal mice. J Comp Neurol. 2008;511:329–341. - PubMed
-
- Amendola J, Verrier B, Roubertoux P, Durand J. Altered sensorimotor development in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci. 2004;20:2822–2826. - PubMed
-
- Beckman JS, Estévez AG, Crow JP, Barbeito L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001;24:S15–S20. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
