

Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates
- PMID: 22742743
- PMCID: PMC3397259
- DOI: 10.1016/j.ajhg.2012.05.022
Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates
Abstract
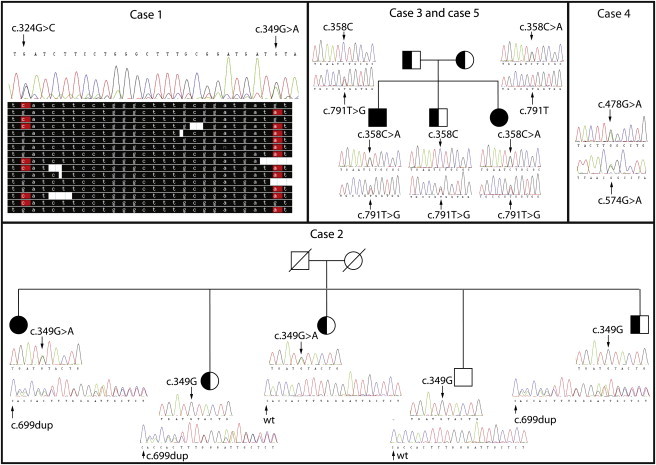
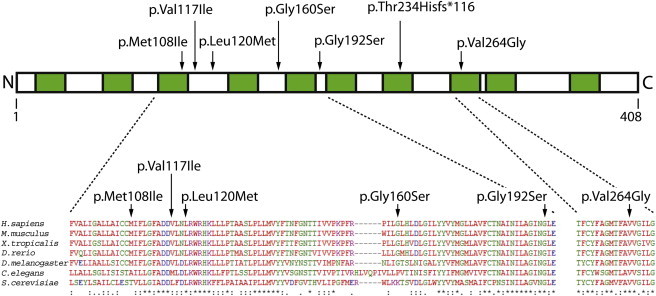
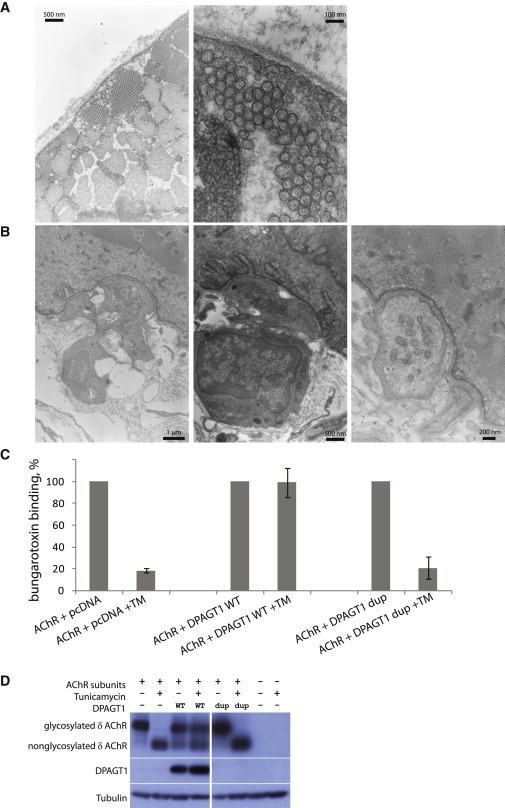
Congenital myasthenic syndromes are a heterogeneous group of inherited disorders that arise from impaired signal transmission at the neuromuscular synapse. They are characterized by fatigable muscle weakness. We performed whole-exome sequencing to determine the underlying defect in a group of individuals with an inherited limb-girdle pattern of myasthenic weakness. We identify DPAGT1 as a gene in which mutations cause a congenital myasthenic syndrome. We describe seven different mutations found in five individuals with DPAGT1 mutations. The affected individuals share a number of common clinical features, including involvement of proximal limb muscles, response to treatment with cholinesterase inhibitors and 3,4-diaminopyridine, and the presence of tubular aggregates in muscle biopsies. Analyses of motor endplates from two of the individuals demonstrate a severe reduction of endplate acetylcholine receptors. DPAGT1 is an essential enzyme catalyzing the first committed step of N-linked protein glycosylation. Our findings underscore the importance of N-linked protein glycosylation for proper functioning of the neuromuscular junction. Using the DPAGT1-specific inhibitor tunicamycin, we show that DPAGT1 is required for efficient glycosylation of acetylcholine-receptor subunits and for efficient export of acetylcholine receptors to the cell surface. We suggest that the primary pathogenic mechanism of DPAGT1 mutations is reduced levels of acetylcholine receptors at the endplate region. These individuals share clinical features similar to those of congenital myasthenic syndrome due to GFPT1 mutations, and their disorder might be part of a larger subgroup comprising the congenital myasthenic syndromes that result from defects in the N-linked glycosylation pathway and that manifest through impaired neuromuscular transmission.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Chaouch A., Beeson D., Hantaï D., Lochmüller H. 186th ENMC International Workshop: Congenital myasthenic syndromes 24-26 June 2011, Naarden, The Netherlands. Neuromuscul. Disord. 2012;22:566–576. - PubMed
-
- Beeson D., Higuchi O., Palace J., Cossins J., Spearman H., Maxwell S., Newsom-Davis J., Burke G., Fawcett P., Motomura M. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–1978. - PubMed
-
- Palace J., Lashley D., Newsom-Davis J., Cossins J., Maxwell S., Kennett R., Jayawant S., Yamanashi Y., Beeson D. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain. 2007;130:1507–1515. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
