

Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA
- PMID: 22744790
- PMCID: PMC3725314
- DOI: 10.1007/s00401-012-1002-8
Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA
Abstract
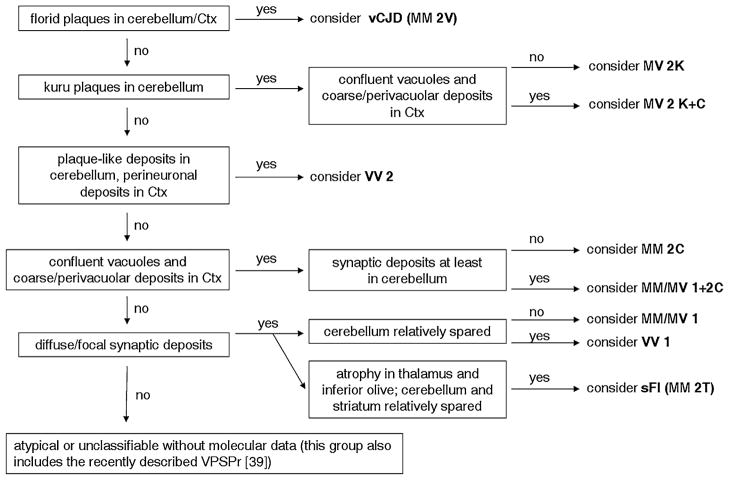
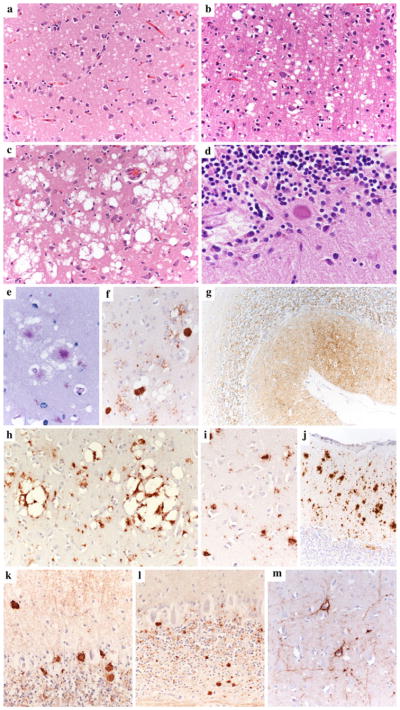
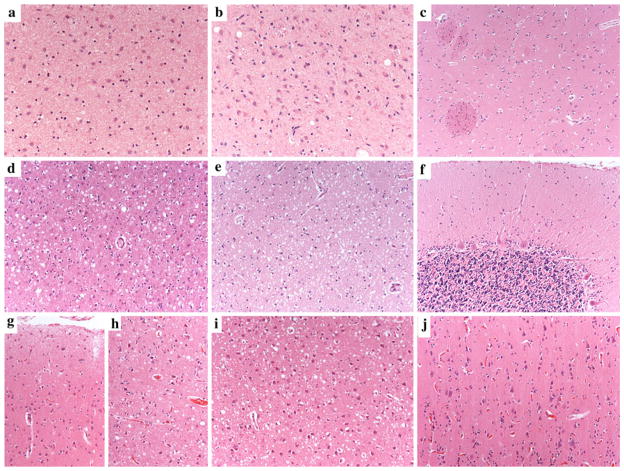
The current classification of human sporadic prion diseases recognizes six major phenotypic subtypes with distinctive clinicopathological features, which largely correlate at the molecular level with the genotype at the polymorphic codon 129 (methionine, M, or valine, V) in the prion protein gene and with the size of the protease-resistant core of the abnormal prion protein, PrP(Sc) (i.e. type 1 migrating at 21 kDa and type 2 at 19 kDa). We previously demonstrated that PrP(Sc) typing by Western blotting is a reliable means of strain typing and disease classification. Limitations of this approach, however, particularly in the interlaboratory setting, are the association of PrP(Sc) types 1 or 2 with more than one clinicopathological phenotype, which precludes definitive case classification if not supported by further analysis, and the difficulty of fully recognizing cases with mixed phenotypic features. In this study, we tested the inter-rater reliability of disease classification based only on histopathological criteria. Slides from 21 cases covering the whole phenotypic spectrum of human sporadic prion diseases, and also including two cases of variant Creutzfeldt-Jakob disease (CJD), were distributed blindly to 13 assessors for classification according to given instructions. The results showed good-to-excellent agreement between assessors in the classification of cases. In particular, there was full agreement (100 %) for the two most common sporadic CJD subtypes and variant CJD, and very high concordance in general for all pure phenotypes and the most common subtype with mixed phenotypic features. The present data fully support the basis for the current classification of sporadic human prion diseases and indicate that, besides molecular PrP(Sc) typing, histopathological analysis permits reliable disease classification with high interlaboratory accuracy.
Figures
References
-
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8:552–561. - PubMed
-
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501. - PubMed
-
- Bruce ME, Boyle A, Cousens S, McConnell I, Foster J, Goldmann W, Fraser H. Strain characterization of natural sheep scrapie and comparison with BSE. J Gen Virol. 2002;83:695–704. - PubMed
-
- Cali I, Castellani R, Alshekhlee A, Cohen Y, Blevins J, Yuan J, Langeveld JP, Parchi P, Safar JG, Zou WQ, Gambetti P. Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt–Jakob disease: its effect on the phenotype and prion-type characteristics. Brain. 2009;132:2643–2658. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
