

Modulation of hERG potassium channel gating normalizes action potential duration prolonged by dysfunctional KCNQ1 potassium channel
- PMID: 22745159
- PMCID: PMC3406814
- DOI: 10.1073/pnas.1205266109
Modulation of hERG potassium channel gating normalizes action potential duration prolonged by dysfunctional KCNQ1 potassium channel
Abstract
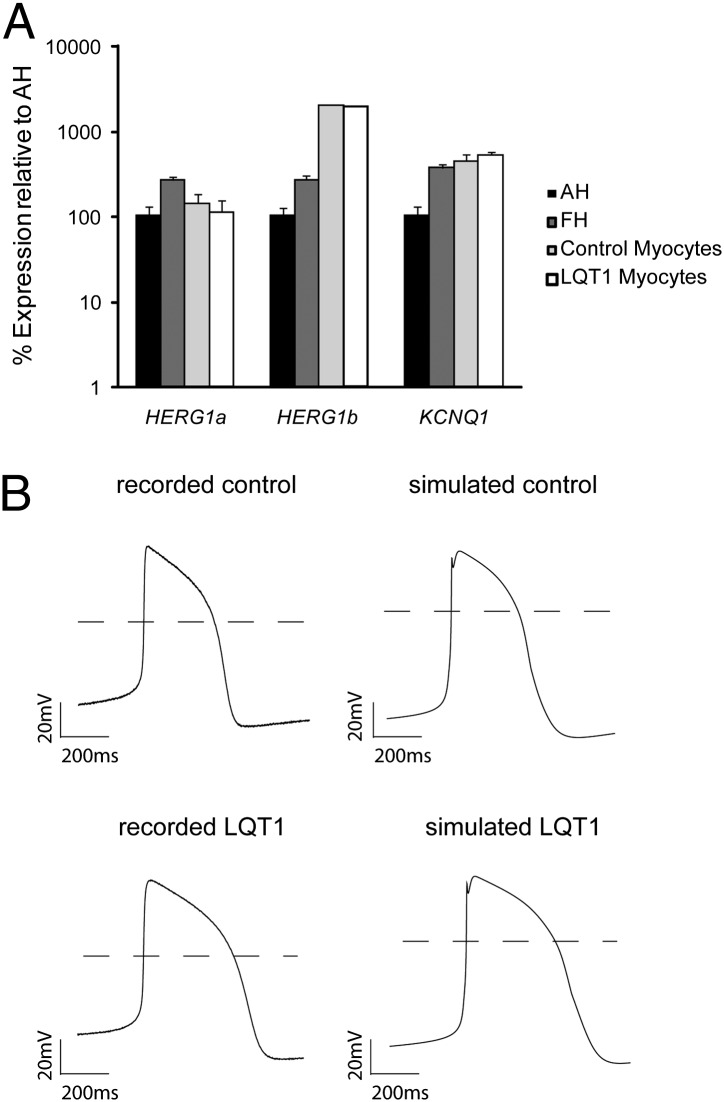
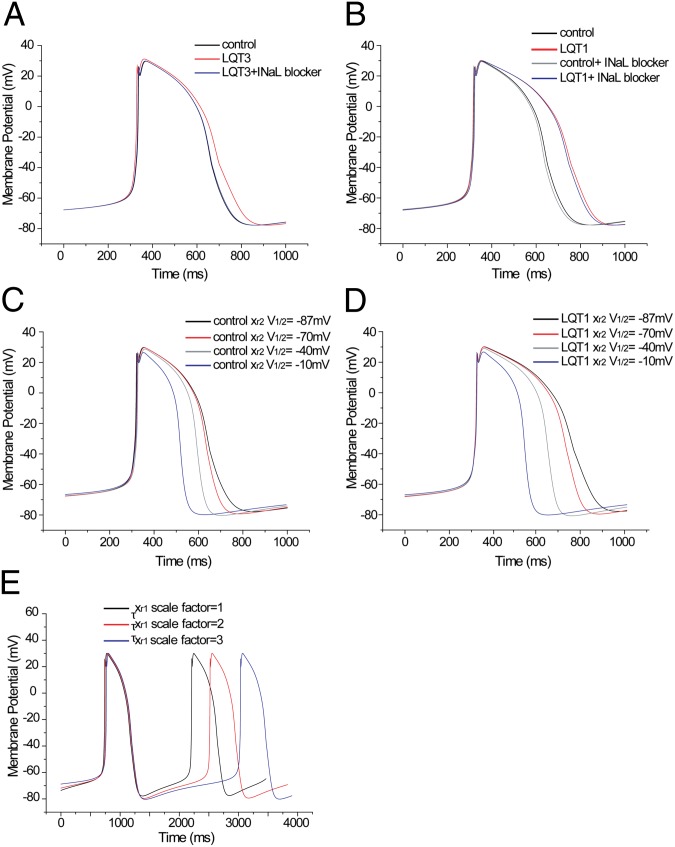
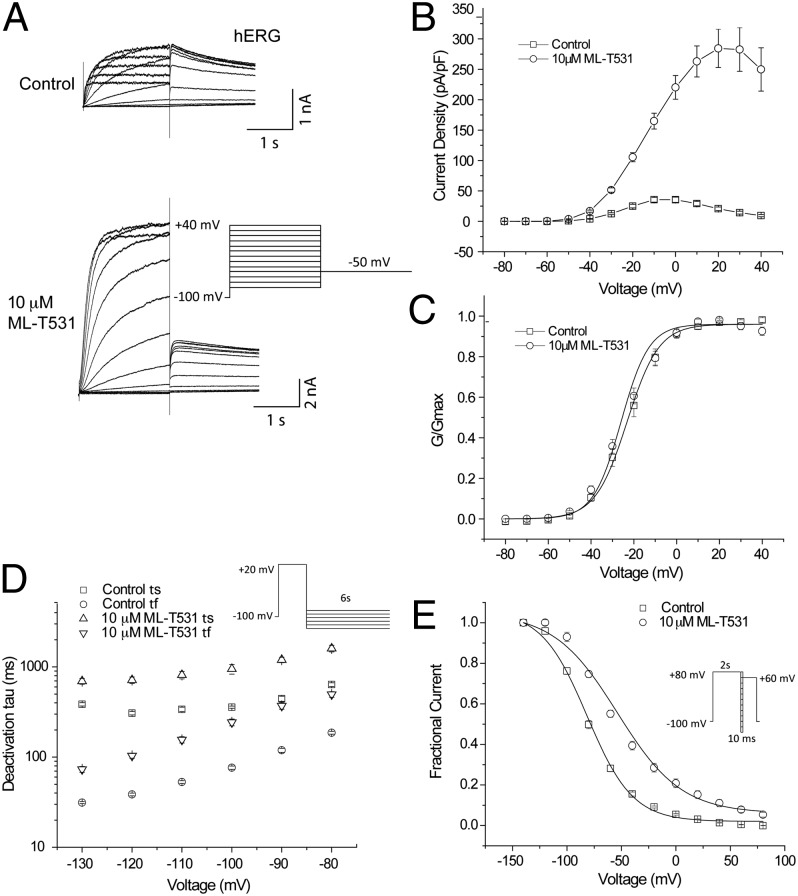
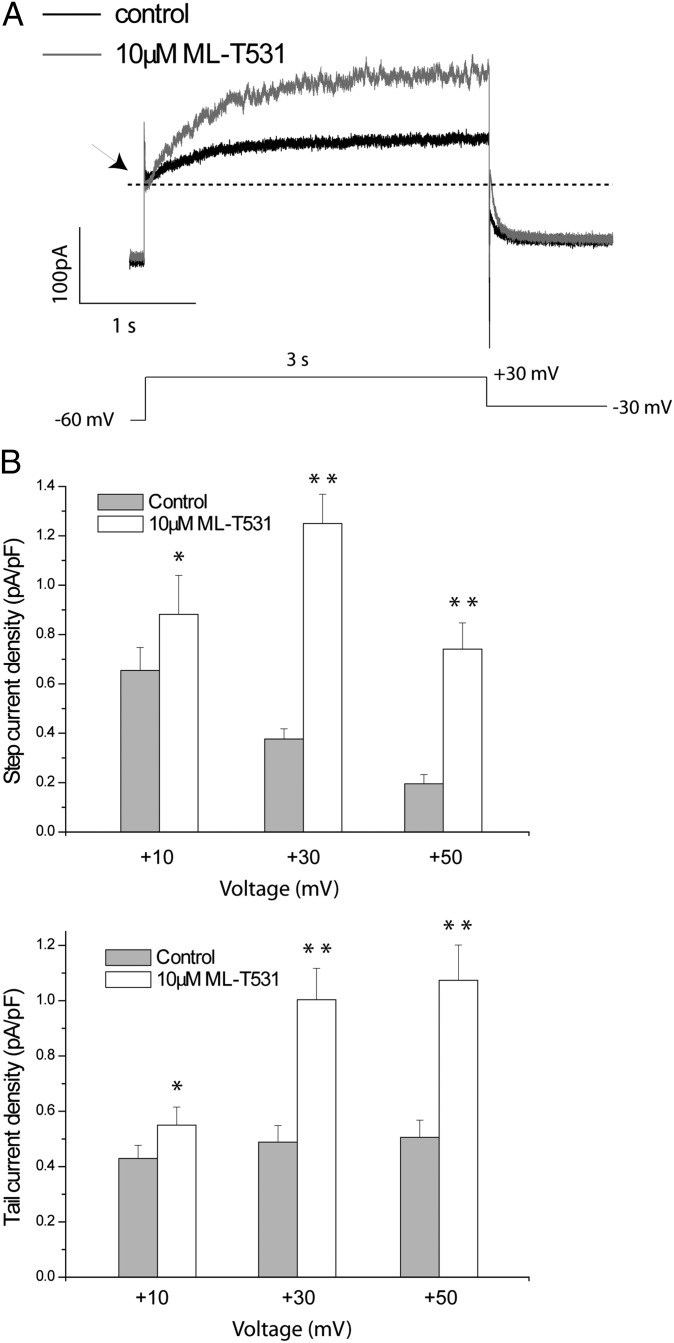
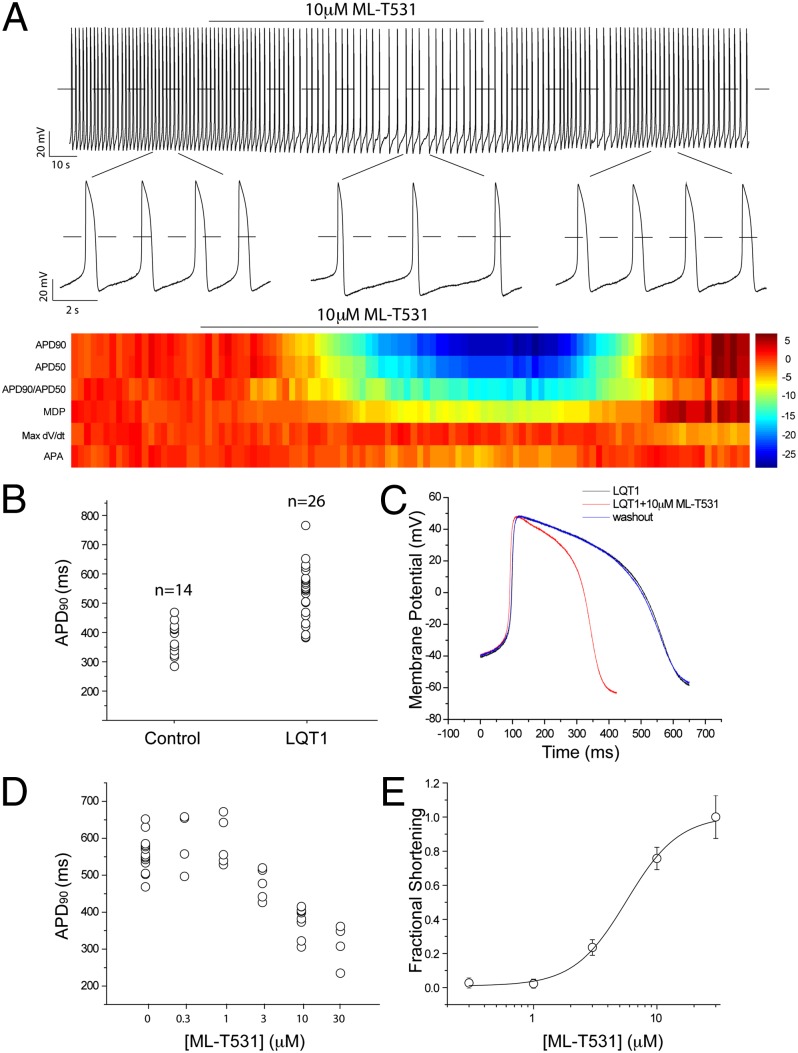
Long QT syndrome (LQTS) is a genetic disease characterized by a prolonged QT interval in an electrocardiogram (ECG), leading to higher risk of sudden cardiac death. Among the 12 identified genes causal to heritable LQTS, ∼90% of affected individuals harbor mutations in either KCNQ1 or human ether-a-go-go related genes (hERG), which encode two repolarizing potassium currents known as I(Ks) and I(Kr). The ability to quantitatively assess contributions of different current components is therefore important for investigating disease phenotypes and testing effectiveness of pharmacological modulation. Here we report a quantitative analysis by simulating cardiac action potentials of cultured human cardiomyocytes to match the experimental waveforms of both healthy control and LQT syndrome type 1 (LQT1) action potentials. The quantitative evaluation suggests that elevation of I(Kr) by reducing voltage sensitivity of inactivation, not via slowing of deactivation, could more effectively restore normal QT duration if I(Ks) is reduced. Using a unique specific chemical activator for I(Kr) that has a primary effect of causing a right shift of V(1/2) for inactivation, we then examined the duration changes of autonomous action potentials from differentiated human cardiomyocytes. Indeed, this activator causes dose-dependent shortening of the action potential durations and is able to normalize action potentials of cells of patients with LQT1. In contrast, an I(Kr) chemical activator of primary effects in slowing channel deactivation was not effective in modulating action potential durations. Our studies provide both the theoretical basis and experimental support for compensatory normalization of action potential duration by a pharmacological agent.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Sanguinetti MC. Long QT syndrome: Ionic basis and arrhythmia mechanism in long QT syndrome type 1. J Cardiovasc Electrophysiol. 2000;11:710–712. - PubMed
-
- Sanguinetti MC, Zou A. Molecular physiology of cardiac delayed rectifier K+ channels. Heart Vessels Suppl. 1997;12:170–172. - PubMed
-
- Hedley PL, et al. The genetic basis of long QT and short QT syndromes: A mutation update. Hum Mutat. 2009;30:1486–1511. - PubMed
-
- Nerbonne JM. Studying cardiac arrhythmias in the mouse—a reasonable model for probing mechanisms? Trends Cardiovasc Med. 2004;14:83–93. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
