

nFuse: discovery of complex genomic rearrangements in cancer using high-throughput sequencing
- PMID: 22745232
- PMCID: PMC3483554
- DOI: 10.1101/gr.136572.111
nFuse: discovery of complex genomic rearrangements in cancer using high-throughput sequencing
Abstract
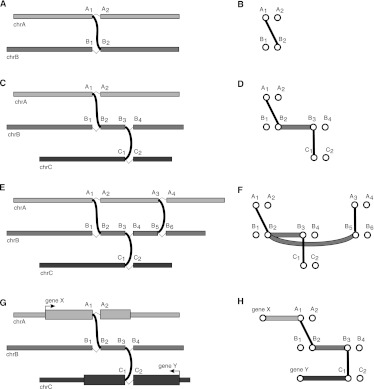
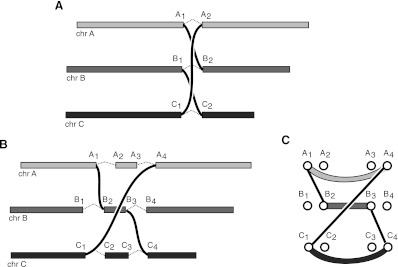
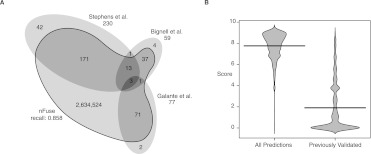
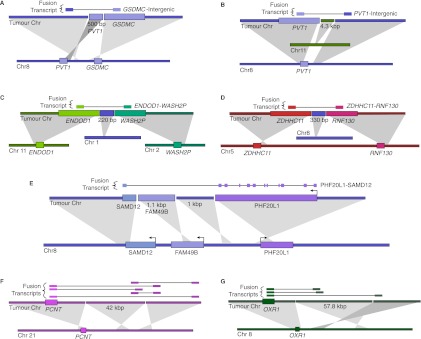
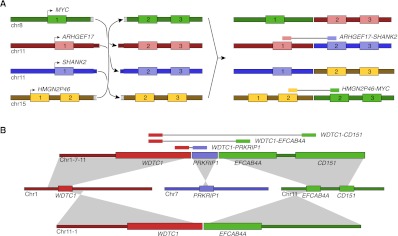
Complex genomic rearrangements (CGRs) are emerging as a new feature of cancer genomes. CGRs are characterized by multiple genomic breakpoints and thus have the potential to simultaneously affect multiple genes, fusing some genes and interrupting other genes. Analysis of high-throughput whole-genome shotgun sequencing (WGSS) is beginning to facilitate the discovery and characterization of CGRs, but further development of computational methods is required. We have developed an algorithmic method for identifying CGRs in WGSS data based on shortest alternating paths in breakpoint graphs. Aiming for a method with the highest possible sensitivity, we use breakpoint graphs built from all WGSS data, including sequences with ambiguous genomic origin. Since the majority of cell function is encoded by the transcriptome, we target our search to find CGRs that underlie fusion transcripts predicted from matched high-throughput cDNA sequencing (RNA-seq). We have applied our method, nFuse, to the discovery of CGRs in publicly available data from the well-studied breast cancer cell line HCC1954 and primary prostate tumor sample 963. We first establish the sensitivity and specificity of the nFuse breakpoint prediction and scoring method using breakpoints previously discovered in HCC1954. We then validate five out of six CGRs in HCC1954 and two out of two CGRs in 963. We show examples of gene fusions that would be difficult to discover using methods that do not account for the existence of CGRs, including one important event that was missed in a previous study of the HCC1954 genome. Finally, we illustrate how CGRs may be used to infer the gene expression history of a tumor.
Figures
References
-
- Asmann YW, Hossain A, Necela BM, Middha S, Kalari KR, Sun Z, Chai HS, Williamson DW, Radisky D, Schroth GP, et al. 2011. A novel bioinformatics pipeline for identification and characterization of fusion transcripts in breast cancer and normal cell lines. Nucleic Acids Res 39: e100 doi: 10.1093/nar/gkr362 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources