

The N-terminal, polybasic region of PrP(C) dictates the efficiency of prion propagation by binding to PrP(Sc)
- PMID: 22745483
- PMCID: PMC3433751
- DOI: 10.1523/JNEUROSCI.1103-12.2012
The N-terminal, polybasic region of PrP(C) dictates the efficiency of prion propagation by binding to PrP(Sc)
Abstract
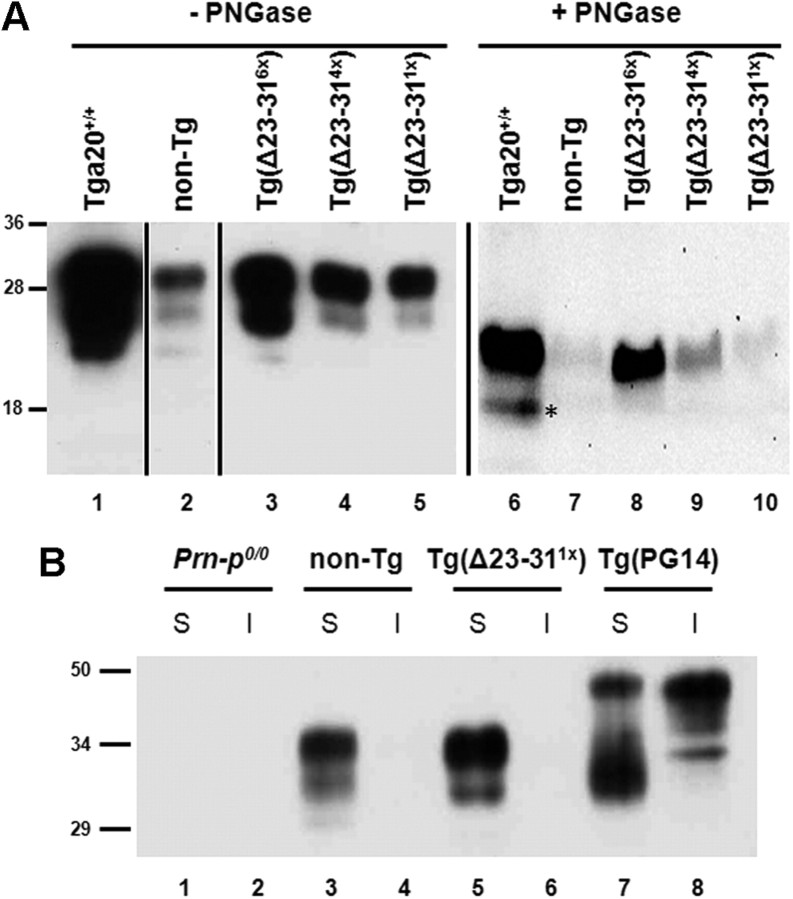
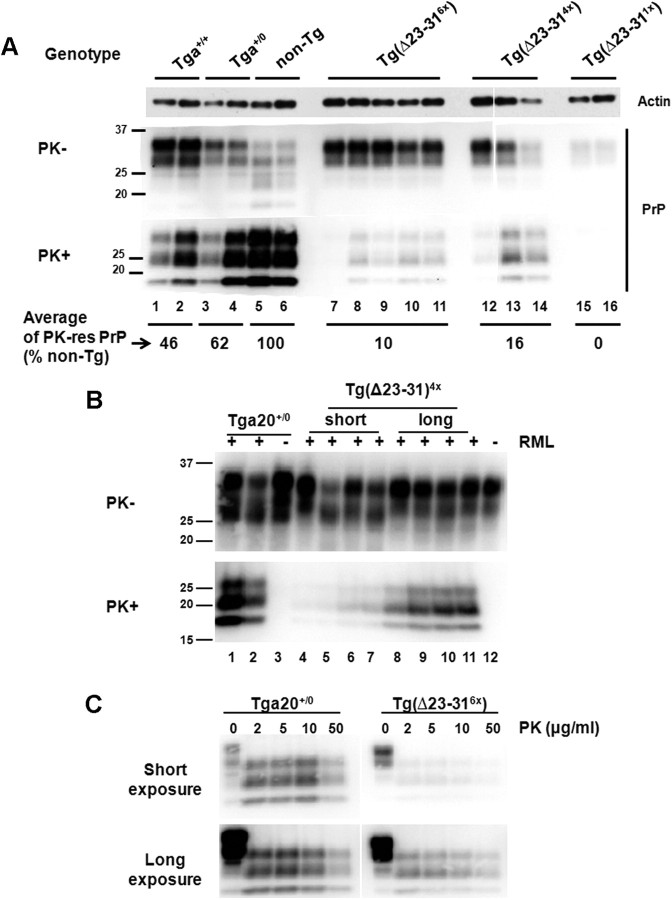
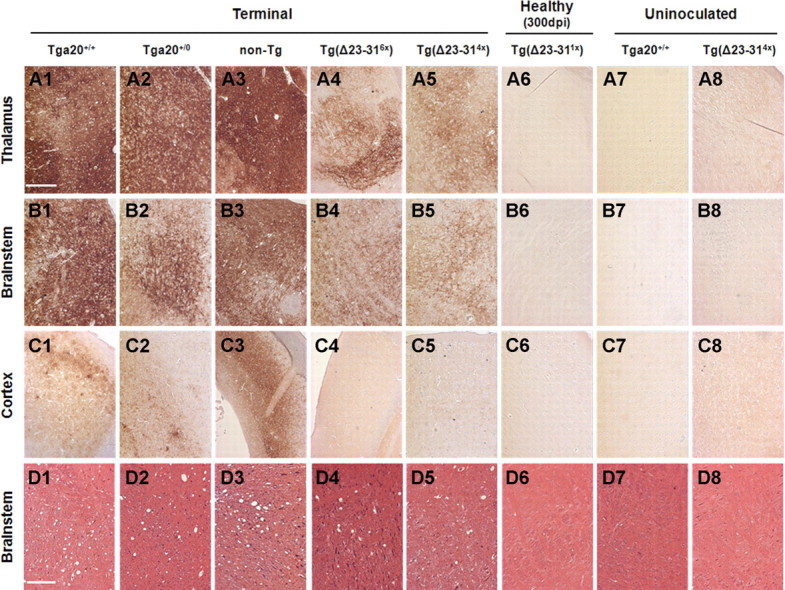
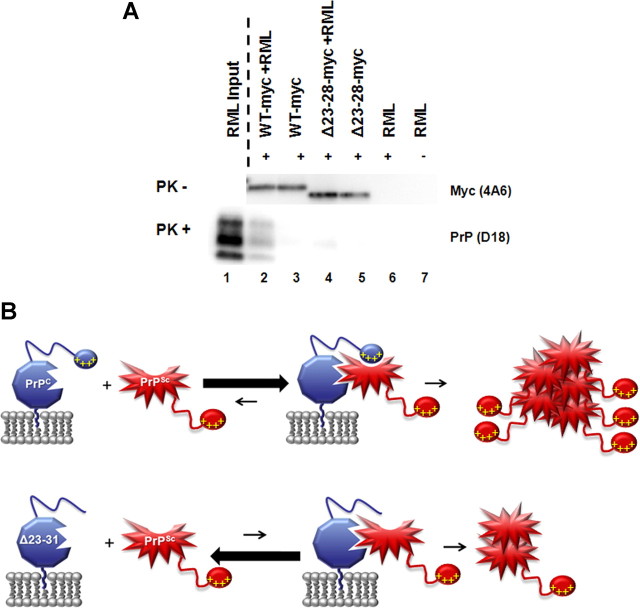
Prion propagation involves a templating reaction in which the infectious form of the prion protein (PrP(Sc)) binds to the cellular form (PrP(C)), generating additional molecules of PrP(Sc). While several regions of the PrP(C) molecule have been suggested to play a role in PrP(Sc) formation based on in vitro studies, the contribution of these regions in vivo is unclear. Here, we report that mice expressing PrP deleted for a short, polybasic region at the N terminus (residues 23-31) display a dramatically reduced susceptibility to prion infection and accumulate greatly reduced levels of PrP(Sc). These results, in combination with biochemical data, demonstrate that residues 23-31 represent a critical site on PrP(C) that binds to PrP(Sc) and is essential for efficient prion propagation. It may be possible to specifically target this region for treatment of prion diseases as well as other neurodegenerative disorders due to β-sheet-rich oligomers that bind to PrP(C).
Figures







References
-
- Bell JE, Gentleman SM, Ironside JW, McCardle L, Lantos PL, Doey L, Lowe J, Fergusson J, Luthert P, McQuaid S, Allen IV. Prion protein immunocytochemistry—UK five centre consensus report. Neuropathol Appl Neurobiol. 1997;23:26–35. - PubMed
-
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials