

Evaluation of the compact high-field orbitrap for top-down proteomics of human cells
- PMID: 22746247
- PMCID: PMC3437942
- DOI: 10.1021/pr3004216
Evaluation of the compact high-field orbitrap for top-down proteomics of human cells
Abstract
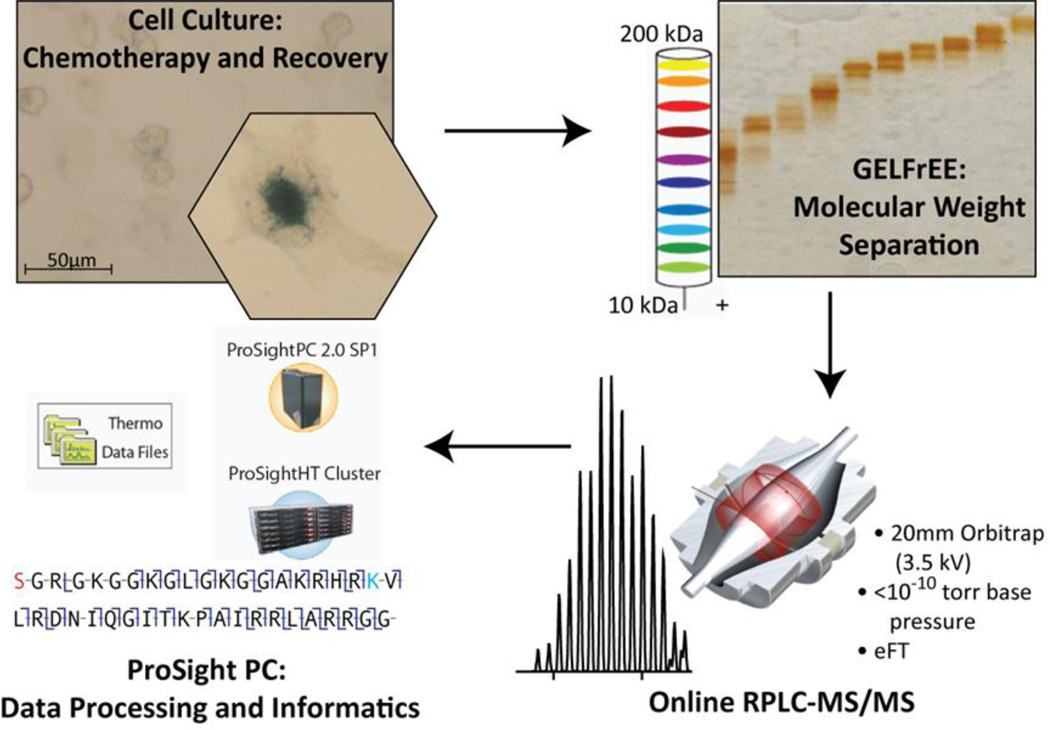
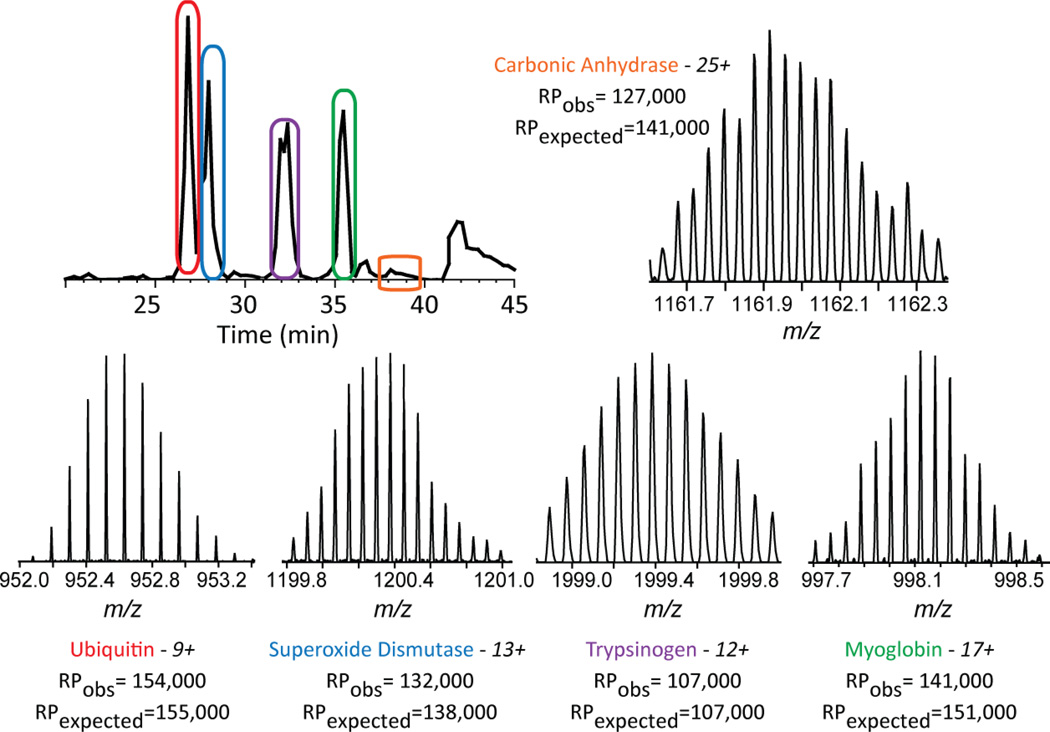
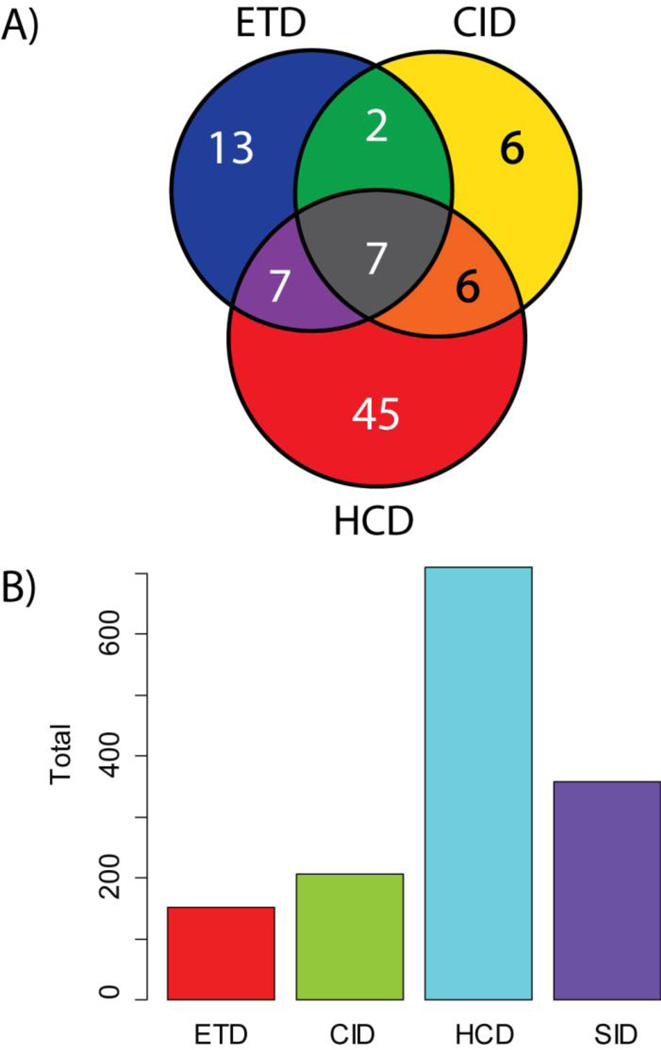
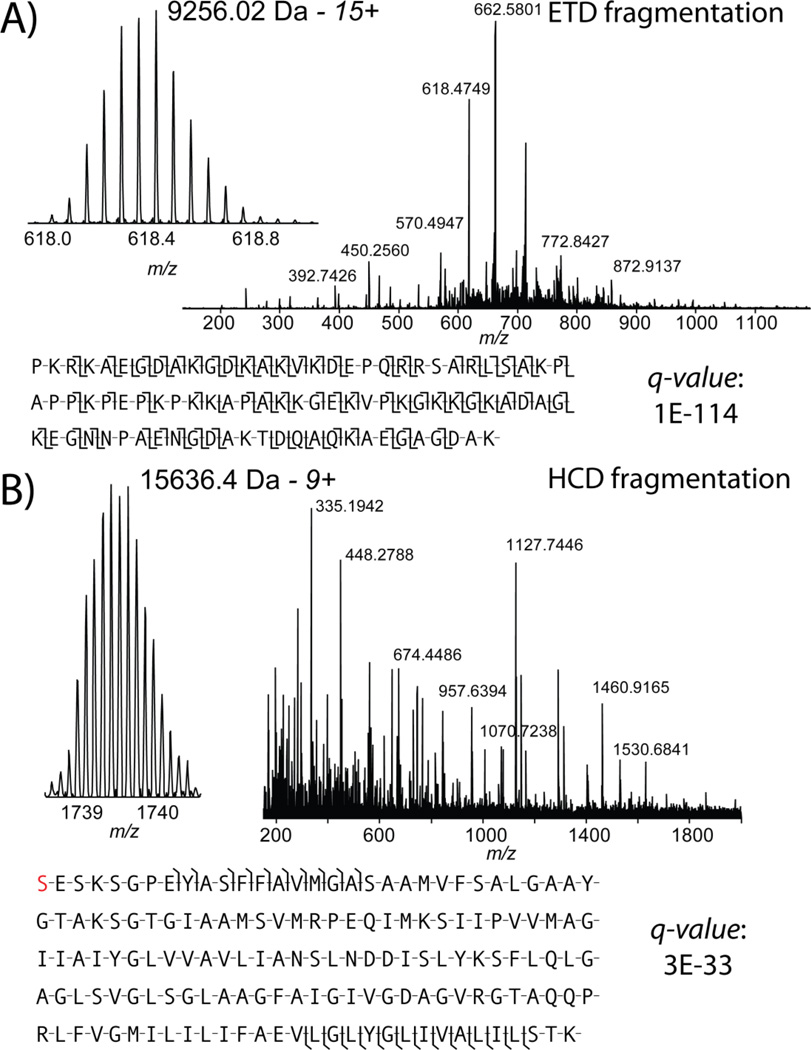
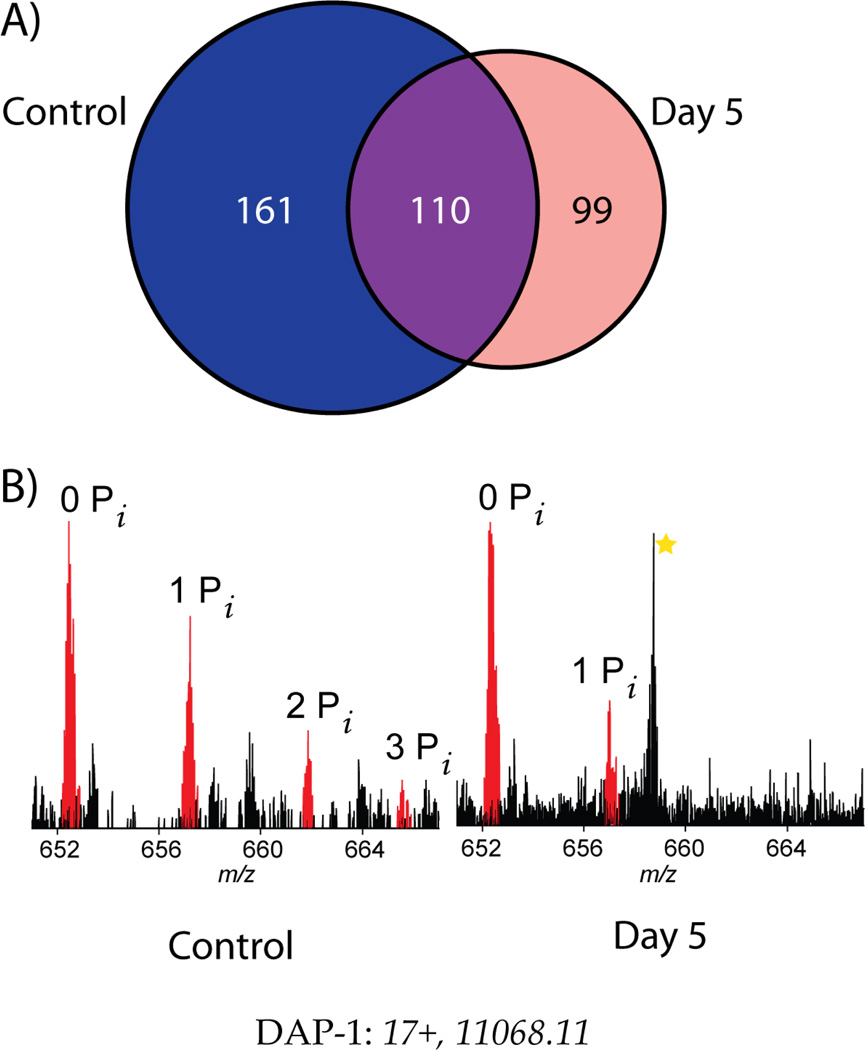
Mass spectrometry based proteomics generally seeks to identify and fully characterize protein species with high accuracy and throughput. Recent improvements in protein separation have greatly expanded the capacity of top-down proteomics (TDP) to identify a large number of intact proteins. To date, TDP has been most tightly associated with Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometry. Here, we couple the improved separations to a Fourier-transform instrument based not on ICR but using the Orbitrap Elite mass analyzer. Application of this platform to H1299 human lung cancer cells resulted in the unambiguous identification of 690 unique proteins and over 2000 proteoforms identified from proteins with intact masses<50 kDa. This is an early demonstration of high throughput TDP (>500 identifications) in an Orbitrap mass spectrometer and exemplifies an accessible platform for whole protein mass spectrometry.
Figures
References
-
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature Methods. 2009;6:359–362. - PubMed
-
- Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SMM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480:254–258. - PMC - PubMed
-
- Service RF. Proteomics ponders prime time. Science. 2008;321:1758–1761. - PubMed
-
- Nesvizhskii A, Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Molecular & Cellular Proteomics. 2005;4:1419–1440. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
