

TAPDANCE: an automated tool to identify and annotate transposon insertion CISs and associations between CISs from next generation sequence data
- PMID: 22748055
- PMCID: PMC3461456
- DOI: 10.1186/1471-2105-13-154
TAPDANCE: an automated tool to identify and annotate transposon insertion CISs and associations between CISs from next generation sequence data
Abstract
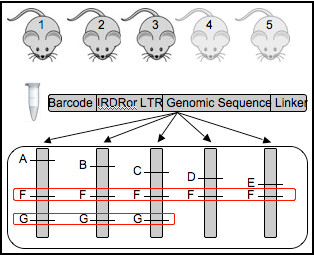
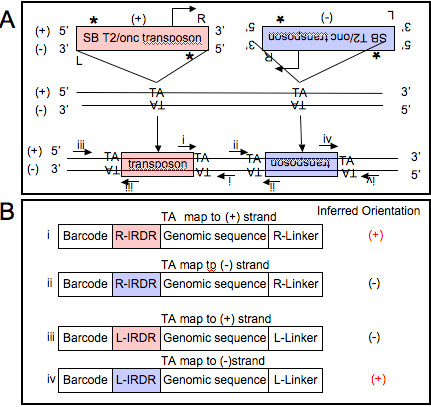
Background: Next generation sequencing approaches applied to the analyses of transposon insertion junction fragments generated in high throughput forward genetic screens has created the need for clear informatics and statistical approaches to deal with the massive amount of data currently being generated. Previous approaches utilized to 1) map junction fragments within the genome and 2) identify Common Insertion Sites (CISs) within the genome are not practical due to the volume of data generated by current sequencing technologies. Previous approaches applied to this problem also required significant manual annotation.
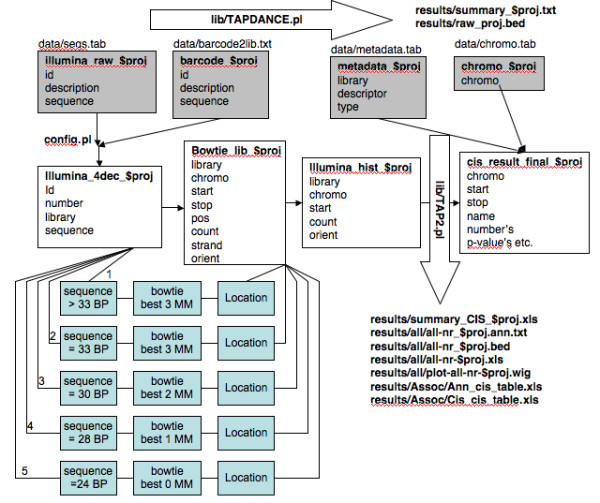
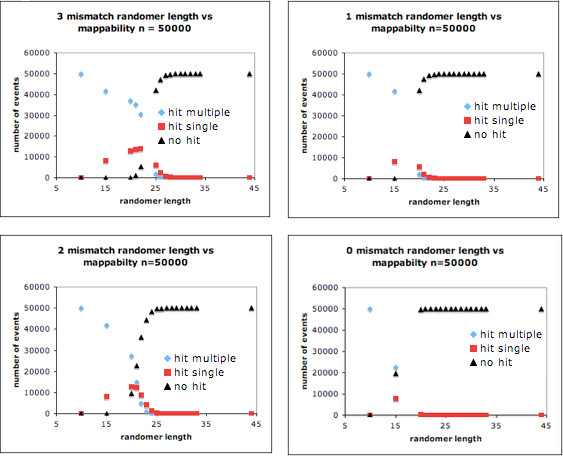
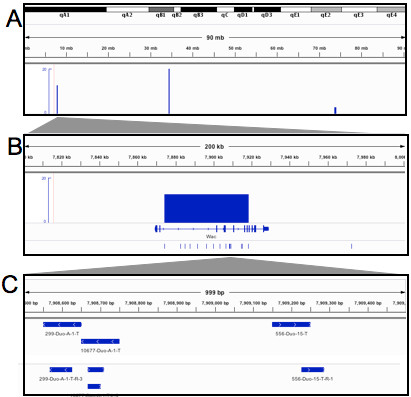
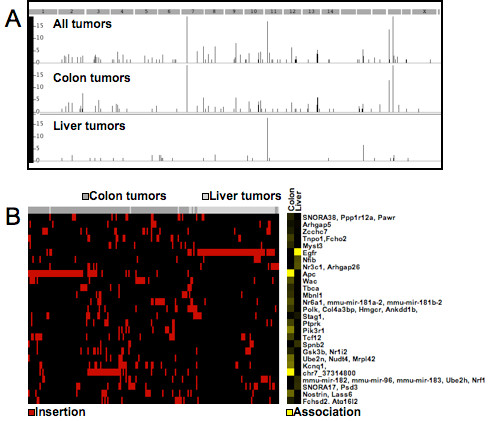
Results: We describe Transposon Annotation Poisson Distribution Association Network Connectivity Environment (TAPDANCE) software, which automates the identification of CISs within transposon junction fragment insertion data. Starting with barcoded sequence data, the software identifies and trims sequences and maps putative genomic sequence to a reference genome using the bowtie short read mapper. Poisson distribution statistics are then applied to assess and rank genomic regions showing significant enrichment for transposon insertion. Novel methods of counting insertions are used to ensure that the results presented have the expected characteristics of informative CISs. A persistent mySQL database is generated and utilized to keep track of sequences, mappings and common insertion sites. Additionally, associations between phenotypes and CISs are also identified using Fisher's exact test with multiple testing correction. In a case study using previously published data we show that the TAPDANCE software identifies CISs as previously described, prioritizes them based on p-value, allows holistic visualization of the data within genome browser software and identifies relationships present in the structure of the data.
Conclusions: The TAPDANCE process is fully automated, performs similarly to previous labor intensive approaches, provides consistent results at a wide range of sequence sampling depth, has the capability of handling extremely large datasets, enables meaningful comparison across datasets and enables large scale meta-analyses of junction fragment data. The TAPDANCE software will greatly enhance our ability to analyze these datasets in order to increase our understanding of the genetic basis of cancers.
Figures


References
-
- Starr TK, Allaei R, Silverstein KAT, Staggs RA, Sarver AL, Bergemann TL, Gupta M, O’Sullivan MG, Matise I, Dupuy AJ, Collier LS, Powers S, Oberg AL, Asmann YW, Thibodeau SN, Tessarollo L, Copeland NG, Jenkins NA, Cormier RT, Largaespada DA. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science. 2009;323:1747–1750. doi: 10.1126/science.1163040. - DOI - PMC - PubMed
-
- Keng VW, Villanueva A, Chiang DY, Dupuy AJ, Ryan BJ, Matise I, Silverstein KAT, Sarver A, Starr TK, Akagi K, Tessarollo L, Collier LS, Powers S, Lowe SW, Jenkins NA, Copeland NG, Llovet JM, Largaespada DA. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat. 2009;27:264–274. doi: 10.1038/nbt.1526. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
