

Structural haplotypes and recent evolution of the human 17q21.31 region
- PMID: 22751096
- PMCID: PMC4020351
- DOI: 10.1038/ng.2334
Structural haplotypes and recent evolution of the human 17q21.31 region
Abstract
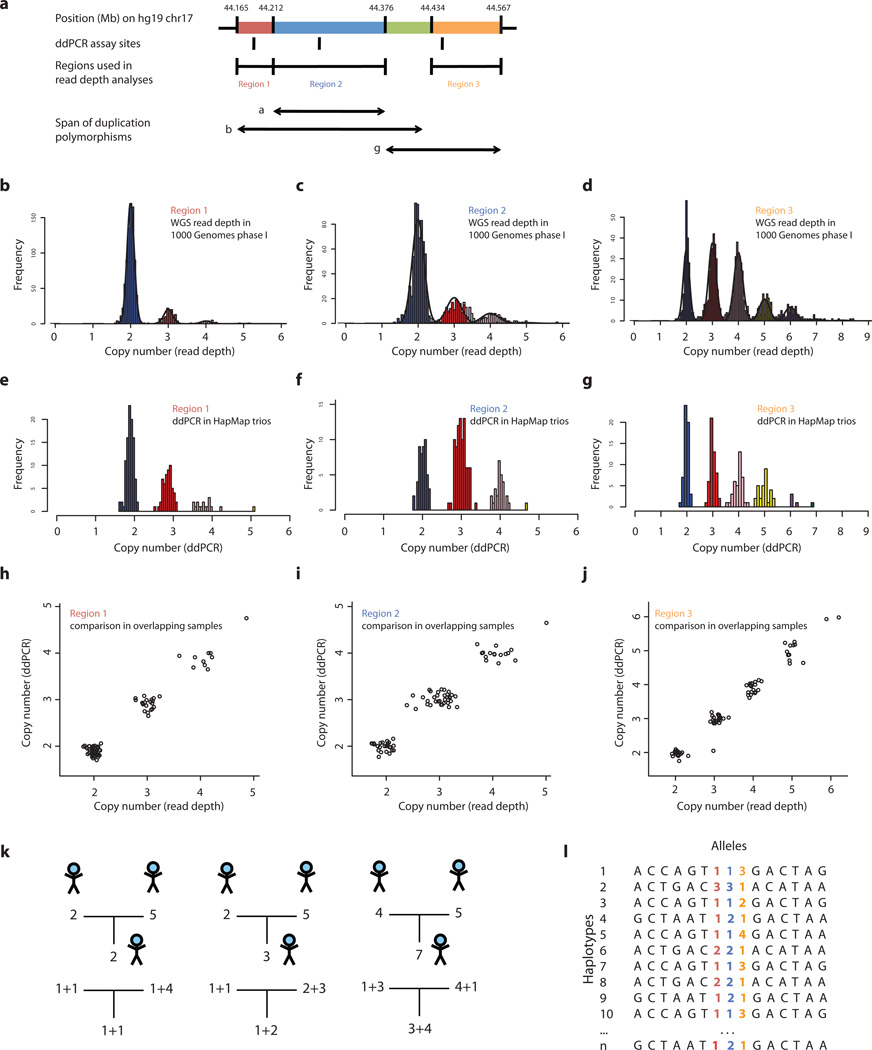
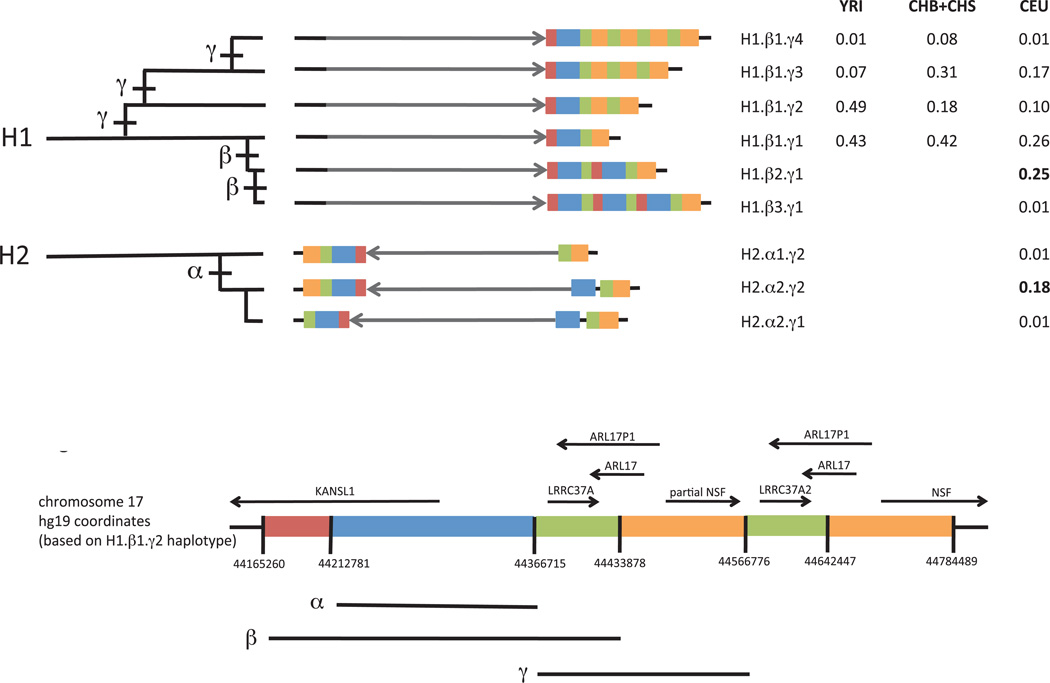
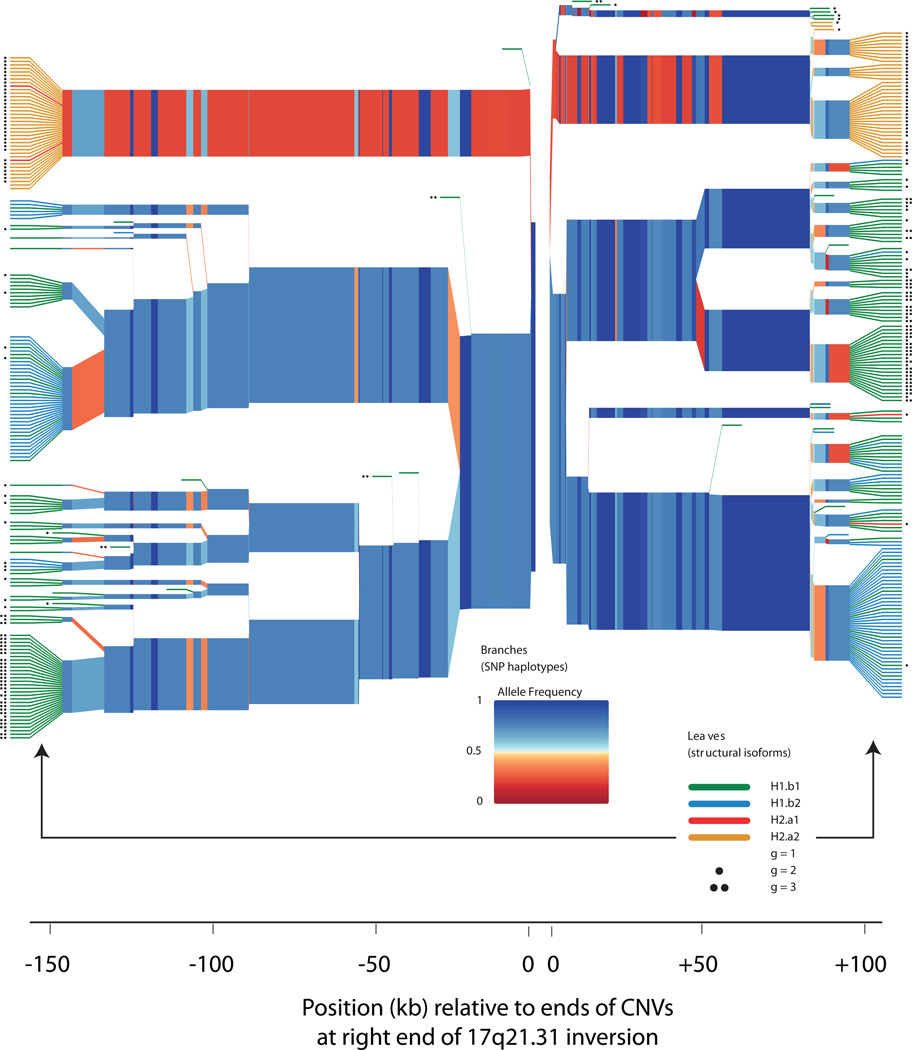
Structurally complex genomic regions are not yet well understood. One such locus, human chromosome 17q21.31, contains a megabase-long inversion polymorphism, many uncharacterized copy-number variations (CNVs) and markers that associate with female fertility, female meiotic recombination and neurological disease. Additionally, the inverted H2 form of 17q21.31 seems to be positively selected in Europeans. We developed a population genetics approach to analyze complex genome structures and identified nine segregating structural forms of 17q21.31. Both the H1 and H2 forms of the 17q21.31 inversion polymorphism contain independently derived, partial duplications of the KANSL1 gene; these duplications, which produce novel KANSL1 transcripts, have both recently risen to high allele frequencies (26% and 19%) in Europeans. An older H2 form lacking such a duplication is present at low frequency in European and central African hunter-gatherer populations. We further show that complex genome structures can be analyzed by imputation from SNPs.
Figures
References
-
- Stefansson H, et al. A common inversion under selection in Europeans. Nat Genet. 2005;37:129–137. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
