

Role of AMPK in UVB-induced DNA damage repair and growth control
- PMID: 22751115
- PMCID: PMC3465498
- DOI: 10.1038/onc.2012.279
Role of AMPK in UVB-induced DNA damage repair and growth control
Abstract
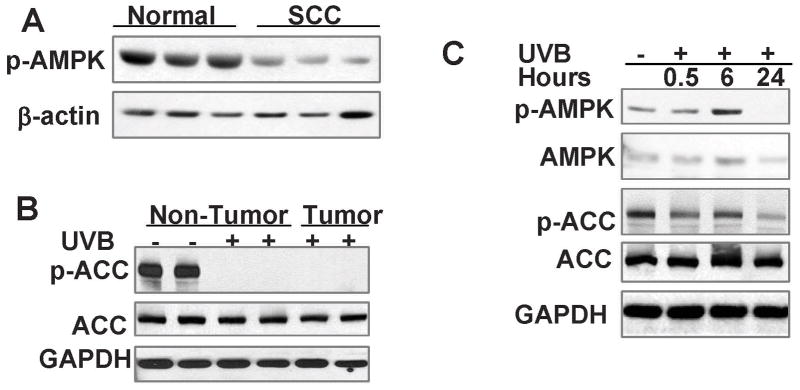
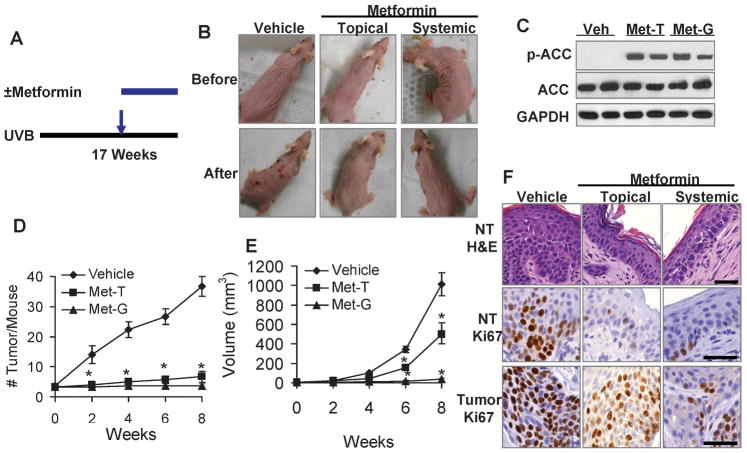
Skin cancer is the most common cancer in the United States, while DNA-damaging ultraviolet B (UVB) radiation from the sun remains the major environmental risk factor. Reducing skin cancer incidence is becoming an urgent issue. The energy-sensing enzyme 5'-AMP-activated protein kinase (AMPK) has a key role in the regulation of cellular lipid and protein metabolism in response to stimuli such as exercise and changes in fuel availability. However, the role of AMPK in the response of skin cells to UVB damage and in skin cancer prevention remains unknown. Here we show that AMPK activation is reduced in human and mouse squamous cell carcinoma as compared with normal skin, and by UVB irradiation, suggesting that AMPK is a tumor suppressor. At the molecular level, AMPK deletion reduced the expression of the DNA repair protein xeroderma pigmentosum C (XPC) and UVB-induced DNA repair. AMPK activation by its activators AICAR (5-aminoimidazole-4-carboxamide ribonucleoside) and metformin (N',N'-dimethylbiguanide), the most widely used antidiabetic drug, increased the expression of XPC and UVB-induced DNA repair in mouse skin, normal human epidermal keratinocytes, and AMPK wild-type (WT) cells but not in AMPK-deficient cells, indicating an AMPK-dependent mechanism. Topical treatment with AICAR and metformin not only delayed onset of UVB-induced skin tumorigenesis but also reduced tumor multiplicity. Furthermore, AMPK deletion increased extracellular signal-regulated kinase (ERK) activation and cell proliferation, whereas AICAR and metformin inhibited ERK activation and cell proliferation in keratinocytes, mouse skin, AMPK WT and AMPK-deficient cells, suggesting an AMPK-independent mechanism. Finally, in UVB-damaged tumor-bearing mice, both topical and systemic metformin prevented the formation of new tumors and suppressed growth of established tumors. Our findings not only suggest that AMPK is a tumor suppressor in the skin by promoting DNA repair and controlling cell proliferation, but also demonstrate previously unknown mechanisms by which the AMPK activators prevent UVB-induced skin tumorigenesis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




Similar articles
-
PTEN positively regulates UVB-induced DNA damage repair.Cancer Res. 2011 Aug 1;71(15):5287-95. doi: 10.1158/0008-5472.CAN-10-4614. Epub 2011 Jul 19. Cancer Res. 2011. PMID: 21771908 Free PMC article.
-
AICAR and metformin, but not exercise, increase muscle glucose transport through AMPK-, ERK-, and PDK1-dependent activation of atypical PKC.Am J Physiol Endocrinol Metab. 2010 Feb;298(2):E179-92. doi: 10.1152/ajpendo.00392.2009. Epub 2009 Nov 3. Am J Physiol Endocrinol Metab. 2010. PMID: 19887597 Free PMC article.
-
AMP-activated protein kinase (AMPK) activation regulates in vitro bone formation and bone mass.Bone. 2010 Aug;47(2):309-19. doi: 10.1016/j.bone.2010.04.596. Epub 2010 Apr 24. Bone. 2010. PMID: 20399918 Free PMC article.
-
AICAr, a Widely Used AMPK Activator with Important AMPK-Independent Effects: A Systematic Review.Cells. 2021 May 4;10(5):1095. doi: 10.3390/cells10051095. Cells. 2021. PMID: 34064363 Free PMC article.
-
UV damage and DNA repair in malignant melanoma and nonmelanoma skin cancer.Adv Exp Med Biol. 2008;624:162-78. doi: 10.1007/978-0-387-77574-6_13. Adv Exp Med Biol. 2008. PMID: 18348455 Review.
Cited by
-
Dysregulation of RNA polymerase I transcription during disease.Biochim Biophys Acta. 2013 Mar-Apr;1829(3-4):342-60. doi: 10.1016/j.bbagrm.2012.10.014. Epub 2012 Nov 12. Biochim Biophys Acta. 2013. PMID: 23153826 Free PMC article. Review.
-
Metformin inhibits skin tumor promotion in overweight and obese mice.Cancer Prev Res (Phila). 2014 Jan;7(1):54-64. doi: 10.1158/1940-6207.CAPR-13-0110. Epub 2013 Nov 6. Cancer Prev Res (Phila). 2014. PMID: 24196830 Free PMC article.
-
Sestrin2 protein positively regulates AKT enzyme signaling and survival in human squamous cell carcinoma and melanoma cells.J Biol Chem. 2014 Dec 26;289(52):35806-14. doi: 10.1074/jbc.M114.595397. Epub 2014 Nov 6. J Biol Chem. 2014. PMID: 25378405 Free PMC article.
-
Mitochondrial dysfunction activates the AMPK signaling and autophagy to promote cell survival.Genes Dis. 2016 Mar;3(1):82-87. doi: 10.1016/j.gendis.2015.12.002. Epub 2016 Jan 4. Genes Dis. 2016. PMID: 28066797 Free PMC article.
-
AMPK: restoring metabolic homeostasis over space and time.Mol Cell. 2021 Sep 16;81(18):3677-3690. doi: 10.1016/j.molcel.2021.08.015. Mol Cell. 2021. PMID: 34547233 Free PMC article. Review.
References
-
- Bode AM, Dong Z. Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci STKE. 2003;2003:RE2. - PubMed
-
- Bowden GT. Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling. Nat Rev Cancer. 2004;4:23–35. - PubMed
-
- Johnson TM, Dolan OM, Hamilton TA, Lu MC, Swanson NA, Lowe L. Clinical and histologic trends of melanoma. J Am Acad Dermatol. 1998;38:681–686. - PubMed
-
- Niggli HJ, Rothlisberger R. Cyclobutane-type pyrimidine photodimer formation and induction of ornithine decarboxylase in human skin fibroblasts after UV irradiation. J Invest Dermatol. 1988;91:579–584. - PubMed
-
- Vink AA, Berg RJ, de Gruijl FR, Roza L, Baan RA. Induction, repair and accumulation of thymine dimers in the skin of UV-B-irradiated hairless mice. Carcinogenesis. 1991;12:861–864. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
