

Particle disease: biologic mechanisms of periprosthetic osteolysis in total hip arthroplasty
- PMID: 22751380
- PMCID: PMC3712274
- DOI: 10.1177/1753425912451779
Particle disease: biologic mechanisms of periprosthetic osteolysis in total hip arthroplasty
Abstract
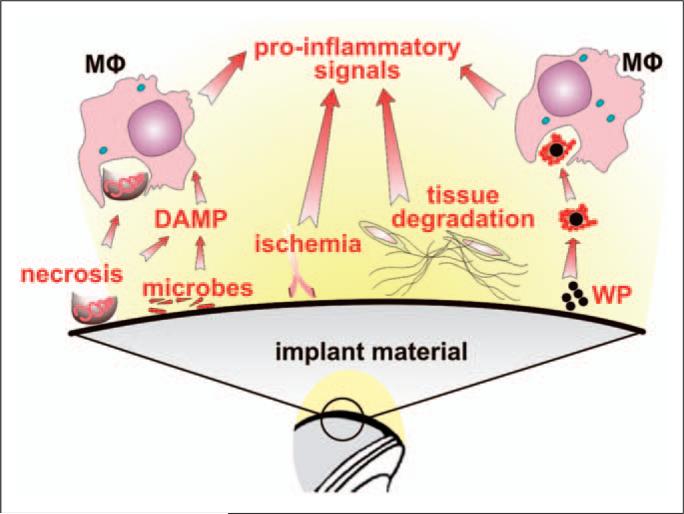
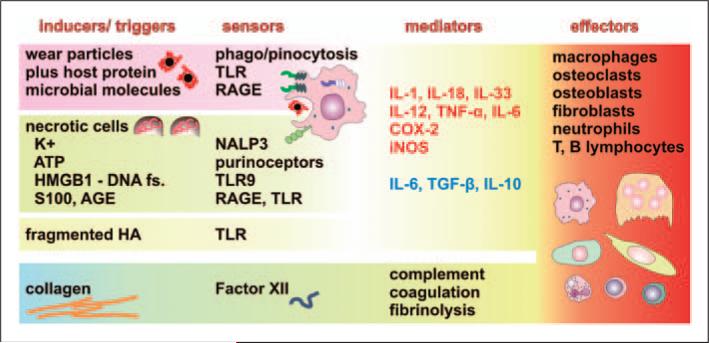
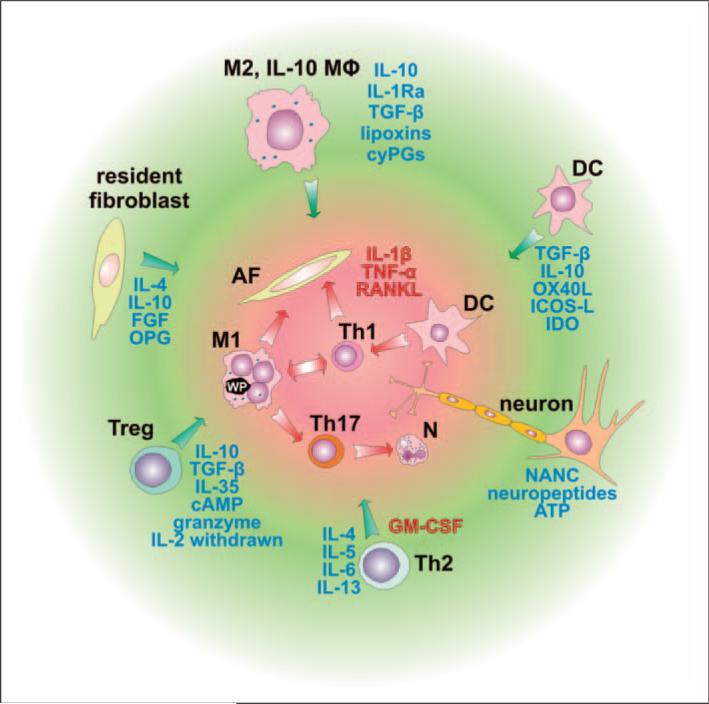
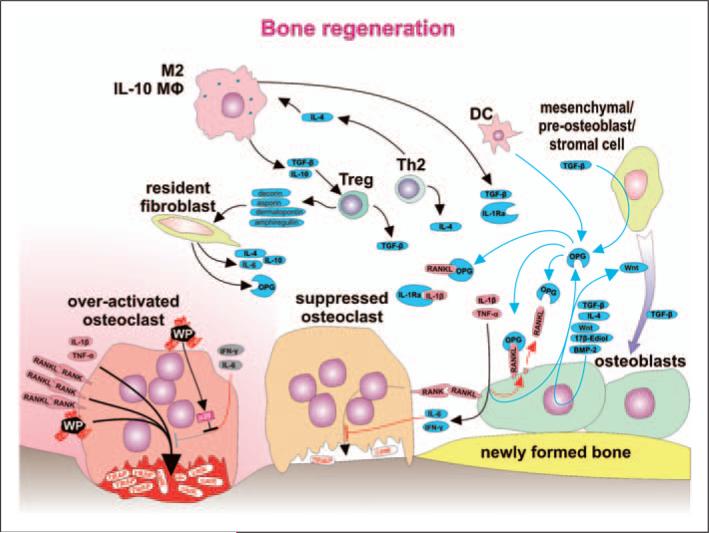
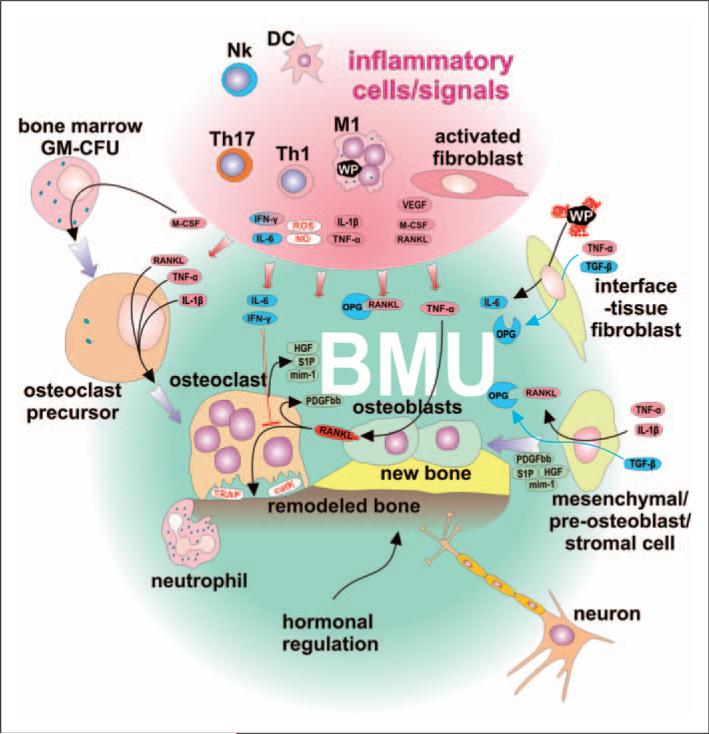
Numerous studies provide detailed insight into the triggering and amplification mechanisms of the inflammatory response associated with prosthetic wear particles, promoting final dominance of bone resorption over bone formation in multiple bone multicellular units around an implant. In fact, inflammation is a highly regulated process tightly linked to simultaneous stimulation of tissue protective and regenerative mechanisms in order to prevent collateral damage of periprosthetic tissues. A variety of cytokines, chemokines, hormones and specific cell populations, including macrophages, dendritic and stem cells, attempt to balance tissue architecture and minimize inflammation. Based on this fact, we postulate that the local tissue homeostatic mechanisms more effectively regulate the pro-inflammatory/pro-osteolytic cells/pathways in patients with none/mild periprosthetic osteolysis (PPOL) than in patients with severe PPOL. In this line of thinking, 'particle disease theory' can be understood, at least partially, in terms of the failure of local tissue homeostatic mechanisms. As a result, we envision focusing current research on homeostatic mechanisms in addition to traditional efforts to elucidate details of pro-inflammatory/pro-osteolytic pathways. We believe this approach could open new avenues for research and potential therapeutic strategies.
Figures
Similar articles
-
Blockade of XCL1/Lymphotactin Ameliorates Severity of Periprosthetic Osteolysis Triggered by Polyethylene-Particles.Front Immunol. 2020 Aug 4;11:1720. doi: 10.3389/fimmu.2020.01720. eCollection 2020. Front Immunol. 2020. PMID: 32849609 Free PMC article.
-
Spinal implant debris-induced osteolysis.Spine (Phila Pa 1976). 2003 Oct 15;28(20):S125-38. doi: 10.1097/00007632-200310151-00006. Spine (Phila Pa 1976). 2003. PMID: 14560184
-
Wear and osteolysis in total joint replacements.Acta Orthop Scand Suppl. 1998 Feb;278:1-16. Acta Orthop Scand Suppl. 1998. PMID: 9524528 Review.
-
Immunobiology of periprosthetic inflammation and pain following ultra-high-molecular-weight-polyethylene wear debris in the lumbar spine.Expert Rev Clin Immunol. 2018 Aug;14(8):695-706. doi: 10.1080/1744666X.2018.1511428. Epub 2018 Aug 21. Expert Rev Clin Immunol. 2018. PMID: 30099915 Free PMC article. Review.
-
Periprosthetic osteolysis: genetics, mechanisms and potential therapeutic interventions.Can J Surg. 2012 Dec;55(6):408-17. doi: 10.1503/cjs.003711. Can J Surg. 2012. PMID: 22992398 Free PMC article. Review.
Cited by
-
Interaction of Materials and Biology in Total Joint Replacement - Successes, Challenges and Future Directions.J Mater Chem B. 2014 Nov 7;2(41):7094-7108. doi: 10.1039/C4TB01005A. J Mater Chem B. 2014. PMID: 25541591 Free PMC article.
-
Long-term survival of the uncemented Balgrist total hip replacement cup.Int Orthop. 2013 Aug;37(8):1449-56. doi: 10.1007/s00264-013-1946-x. Epub 2013 Jun 8. Int Orthop. 2013. PMID: 23744502 Free PMC article.
-
Effect of Aging on the Macrophage Response to Titanium Particles.J Orthop Res. 2020 Feb;38(2):405-416. doi: 10.1002/jor.24461. Epub 2019 Sep 18. J Orthop Res. 2020. PMID: 31498470 Free PMC article.
-
[Particle disease and its effects on periarticular tissue].Orthopadie (Heidelb). 2023 Mar;52(3):196-205. doi: 10.1007/s00132-023-04348-8. Epub 2023 Mar 3. Orthopadie (Heidelb). 2023. PMID: 36867226 Review. German.
-
Effects of metal ions on caspase-1 activation and interleukin-1β release in murine bone marrow-derived macrophages.PLoS One. 2018 Aug 23;13(8):e0199936. doi: 10.1371/journal.pone.0199936. eCollection 2018. PLoS One. 2018. PMID: 30138321 Free PMC article.
References
-
- Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am 2007. 89:780–785. - PubMed
-
- Gallo J, Kaminek P, Zapletalova J, Cechova I, Spicka J, Ditmar R. Is osteolysis associated with a stable total hip replacement asymptomatic? Acta Chir Orthop Traumatol Cech. 2004;71:20–25. - PubMed
-
- Maloney W, Rosenberg A. What is the outcome of treatment for osteolysis? J Am Acad Orthop Surg. 2008;16(Suppl. 1):S26–S32. - PubMed
-
- Deirmengian GK, Zmistowski B, O'Neil JT, Hozack WJ. Management of acetabular bone loss in revision total hip arthroplasty. J Bone Joint Surg Am. 2011;93:1842–1852. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
