

The role of autophagy during coxsackievirus infection of neural progenitor and stem cells
- PMID: 22751470
- PMCID: PMC3427259
- DOI: 10.4161/auto.19781
The role of autophagy during coxsackievirus infection of neural progenitor and stem cells
Abstract
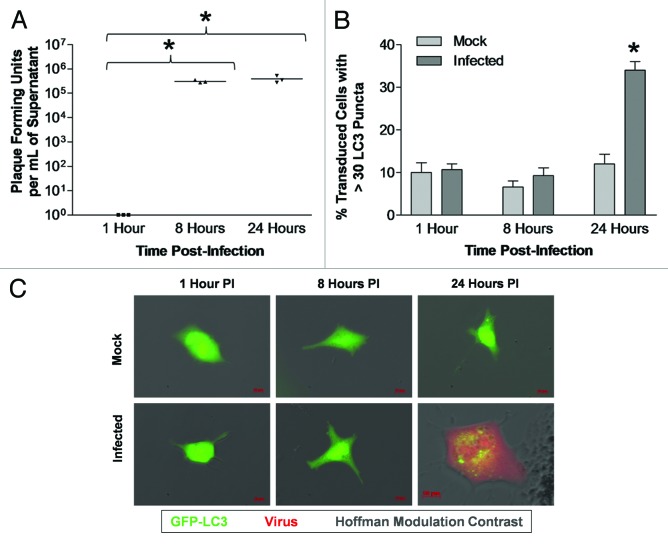
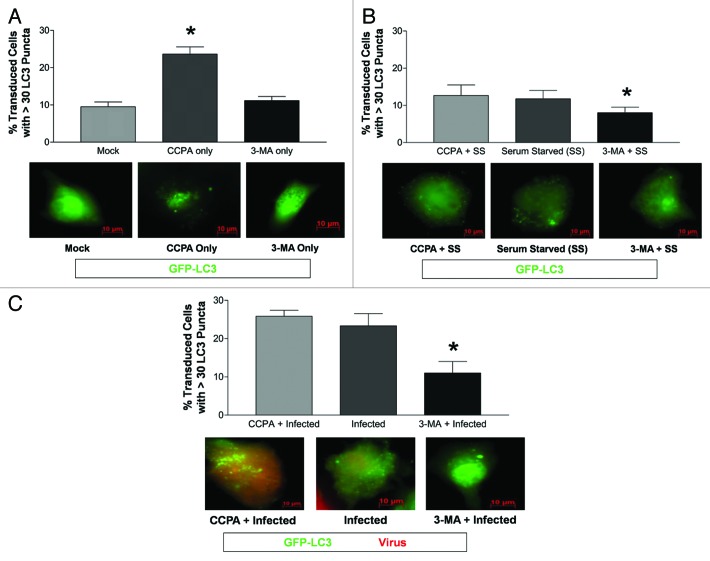
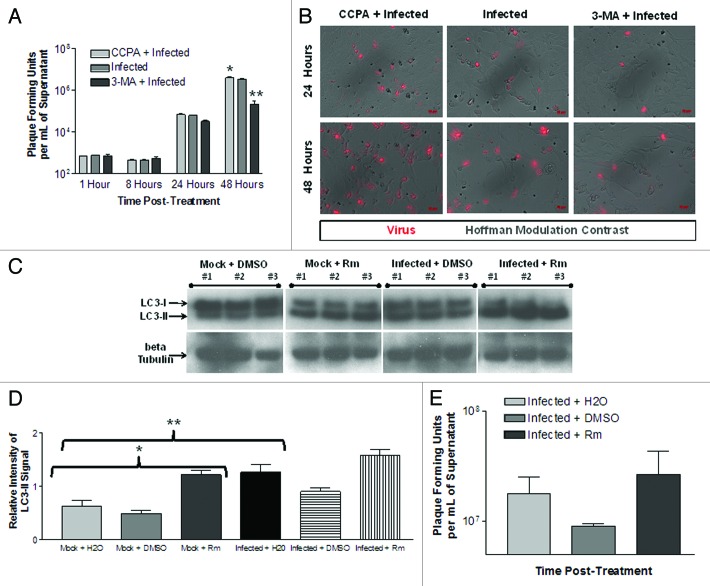
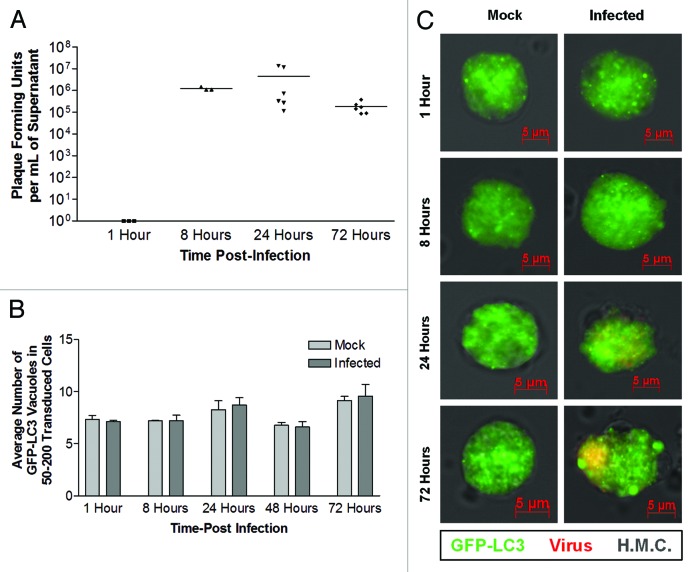
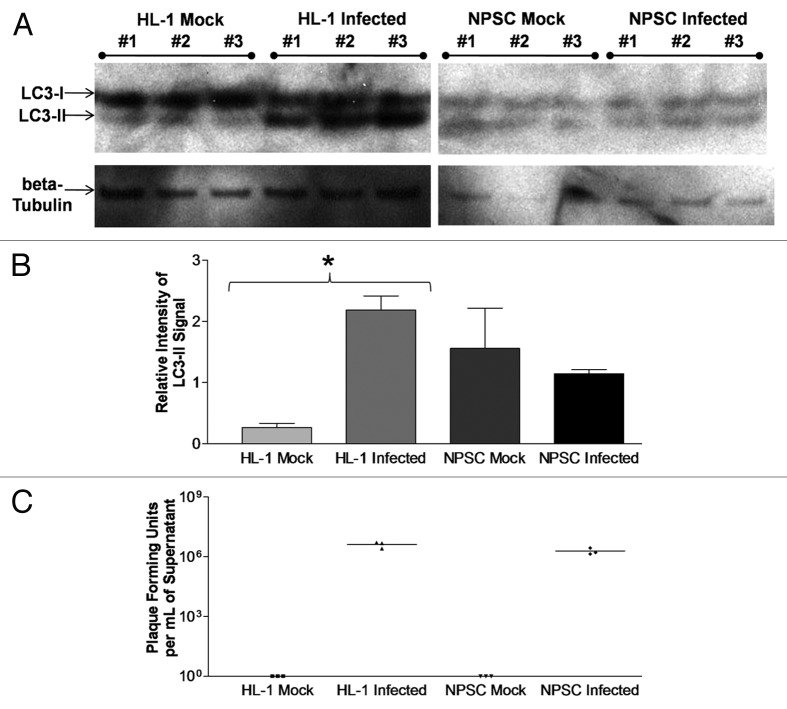
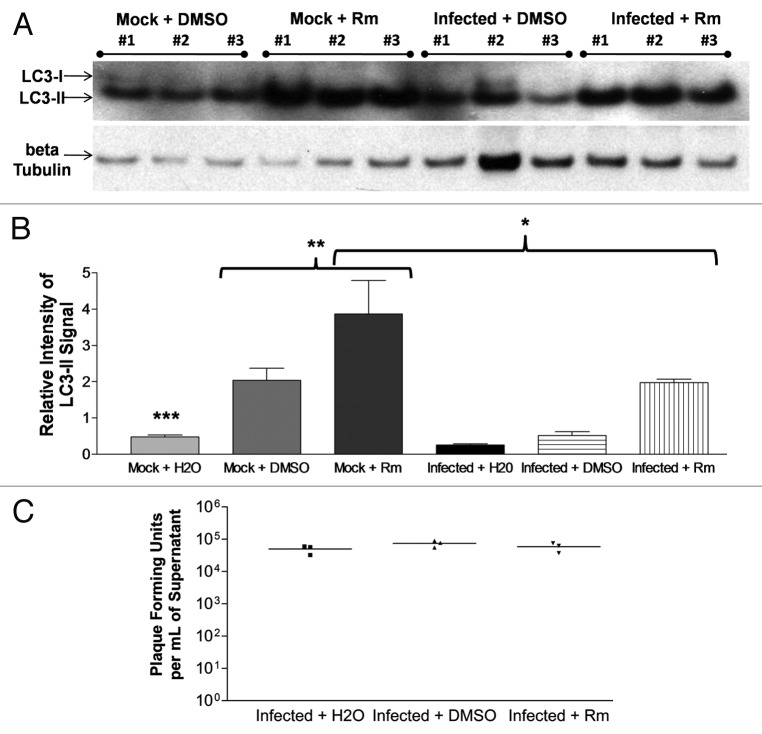
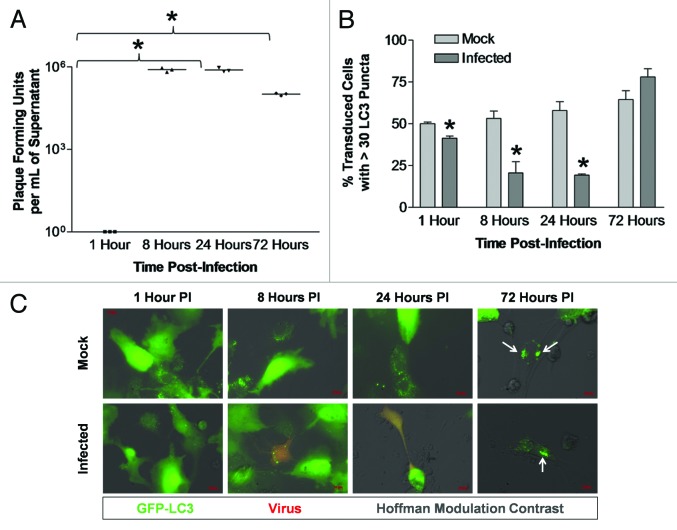
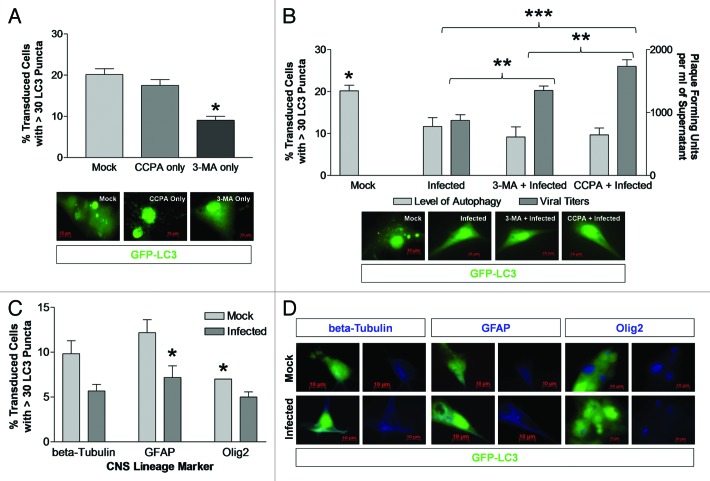
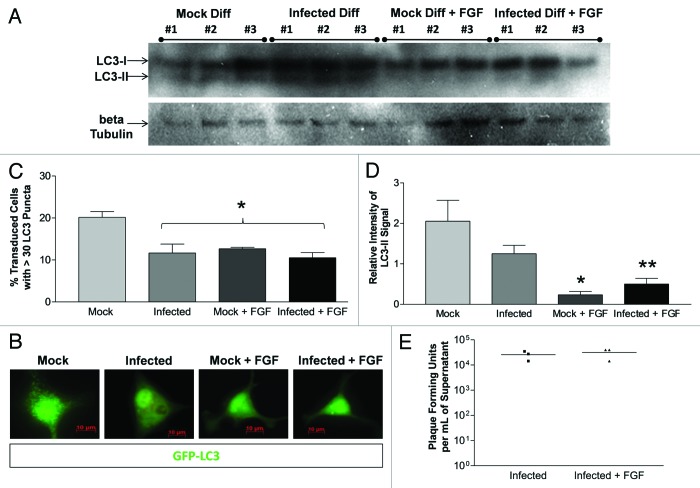
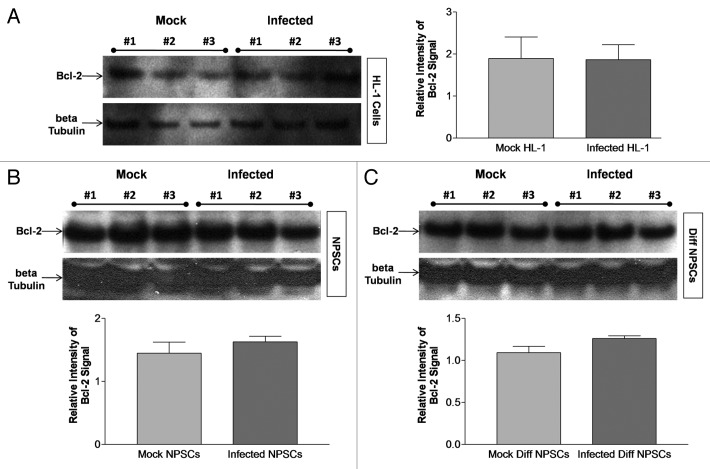
Coxsackievirus B3 (CVB3) has previously been shown to utilize autophagy in an advantageous manner during the course of infection of the host cell. However, few studies have determined whether stem cells induce autophagy in a similar fashion, and whether virus-induced autophagy occurs following infection of stem cells. Therefore, we compared the induction of autophagy following CVB3 infection of neural progenitor and stem cells (NPSCs), which we have recently shown to be highly susceptible to CVB3 infection, to HL-1 cells, a transformed cardiomyocyte cell line. As previously demonstrated for other susceptible host cells, HL-1 cells showed an increase in the activity of autophagic signaling following infection with a CVB3 expressing dsRed protein (dsRed-CVB3). Furthermore, viral titers in HL-1 cells increased in the presence of an inducer of autophagy (CCPA), while viral titers decreased in the presence of an inhibitor of autophagy (3-MA). In contrast, no change in autophagic signaling was seen in NPSCs following infection with dsRed-CVB3. Also, basal levels of autophagy in NPSCs were found to be highly elevated in comparison to HL-1 cells. Autophagy could be induced in NPSCs in the presence of rapamycin without altering levels of dsRed-CVB3 replication. In differentiated NPSC precursors, autophagy was activated during the differentiation process, and a decrease in autophagic signaling was observed within all three CNS lineages following dsRed-CVB3 infection. Hence, we conclude that the role of autophagy in modulating CVB3 replication appears cell type-specific, and stem cells may uniquely regulate autophagy in response to infection.
Figures
Similar articles
-
Coxsackievirus preferentially replicates and induces cytopathic effects in undifferentiated neural progenitor cells.J Virol. 2011 Jun;85(12):5718-32. doi: 10.1128/JVI.02261-10. Epub 2011 Apr 6. J Virol. 2011. PMID: 21471247 Free PMC article.
-
Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers.PLoS Pathog. 2014 Apr 10;10(4):e1004045. doi: 10.1371/journal.ppat.1004045. eCollection 2014 Apr. PLoS Pathog. 2014. PMID: 24722773 Free PMC article.
-
Rapamycin and ZSTK474 can have differential effects at different post‑infection time‑points regarding CVB3 replication and CVB3‑induced autophagy.Mol Med Rep. 2018 Jul;18(1):1088-1094. doi: 10.3892/mmr.2018.9037. Epub 2018 May 17. Mol Med Rep. 2018. PMID: 29845290
-
A Kidnapping Story: How Coxsackievirus B3 and Its Host Cell Interact.Cell Physiol Biochem. 2019;53(1):121-140. doi: 10.33594/000000125. Cell Physiol Biochem. 2019. PMID: 31230428 Review.
-
Coxsackievirus B3 replication and pathogenesis.Future Microbiol. 2015;10(4):629-53. doi: 10.2217/fmb.15.5. Future Microbiol. 2015. PMID: 25865198 Review.
Cited by
-
Beclin1 Binds to Enterovirus 71 3D Protein to Promote the Virus Replication.Viruses. 2020 Jul 14;12(7):756. doi: 10.3390/v12070756. Viruses. 2020. PMID: 32674313 Free PMC article.
-
Exosomes mediate Coxsackievirus B3 transmission and expand the viral tropism.PLoS Pathog. 2023 Jan 12;19(1):e1011090. doi: 10.1371/journal.ppat.1011090. eCollection 2023 Jan. PLoS Pathog. 2023. PMID: 36634130 Free PMC article.
-
The impact of juvenile coxsackievirus infection on cardiac progenitor cells and postnatal heart development.PLoS Pathog. 2014 Jul 31;10(7):e1004249. doi: 10.1371/journal.ppat.1004249. eCollection 2014 Jul. PLoS Pathog. 2014. PMID: 25079373 Free PMC article.
-
Myocardial Damage by SARS-CoV-2: Emerging Mechanisms and Therapies.Viruses. 2021 Sep 21;13(9):1880. doi: 10.3390/v13091880. Viruses. 2021. PMID: 34578462 Free PMC article. Review.
-
A Feedback Loop between TGF-β1 and ATG5 Mediated by miR-122-5p Regulates Fibrosis and EMT in Human Trabecular Meshwork Cells.Curr Issues Mol Biol. 2023 Mar 13;45(3):2381-2392. doi: 10.3390/cimb45030154. Curr Issues Mol Biol. 2023. PMID: 36975524 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials