

Regulation of insulin signaling by the phosphatidylinositol 3,4,5-triphosphate phosphatase SKIP through the scaffolding function of Pak1
- PMID: 22751929
- PMCID: PMC3422014
- DOI: 10.1128/MCB.00636-12
Regulation of insulin signaling by the phosphatidylinositol 3,4,5-triphosphate phosphatase SKIP through the scaffolding function of Pak1
Abstract
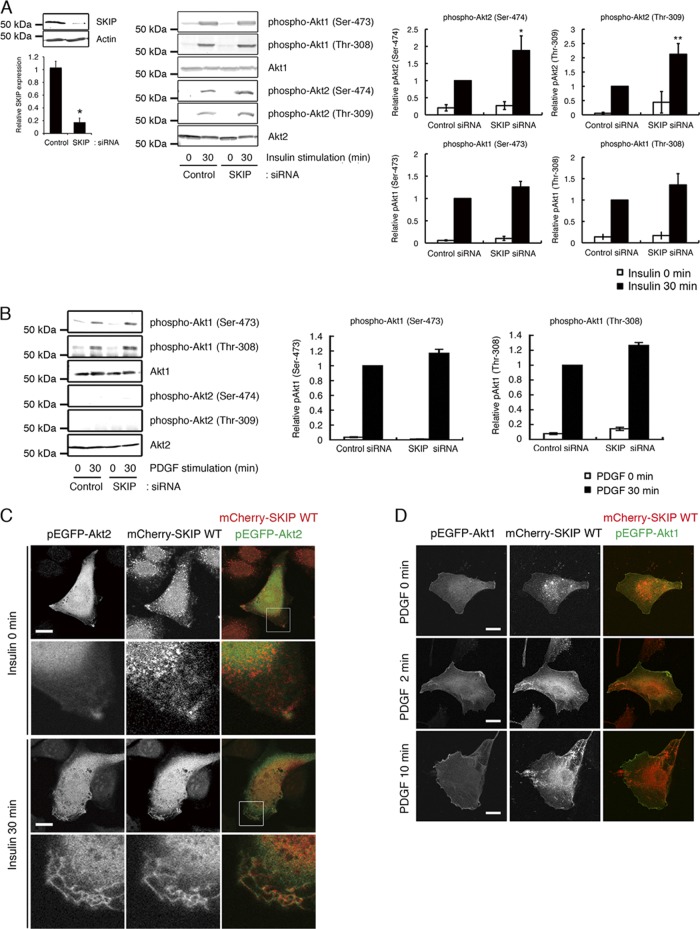
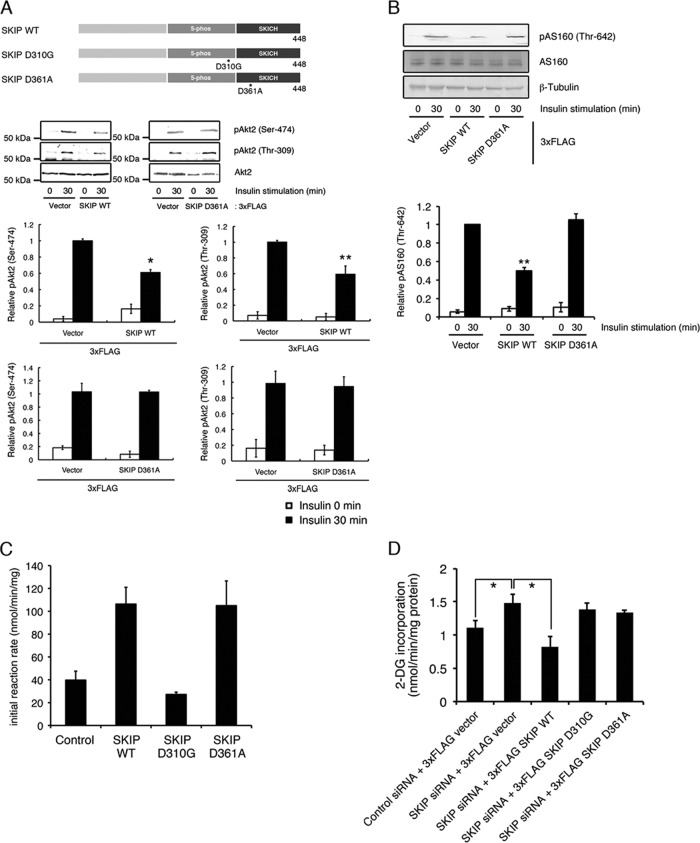
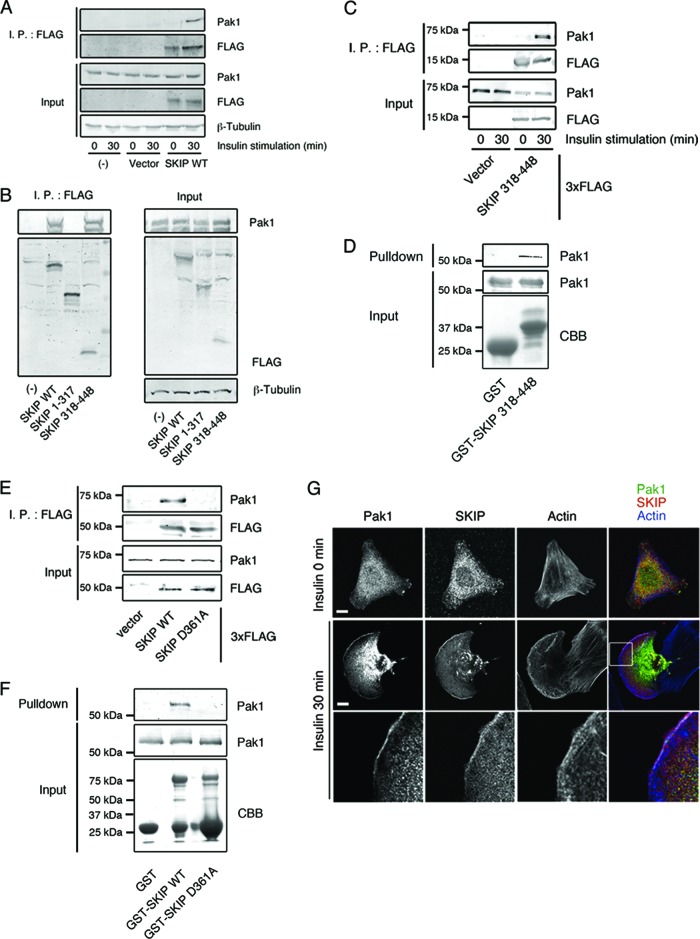
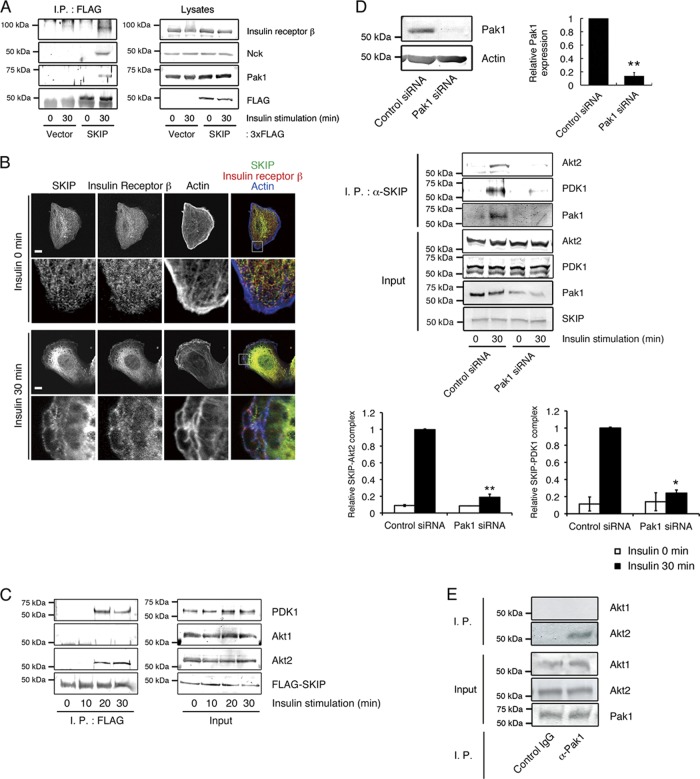
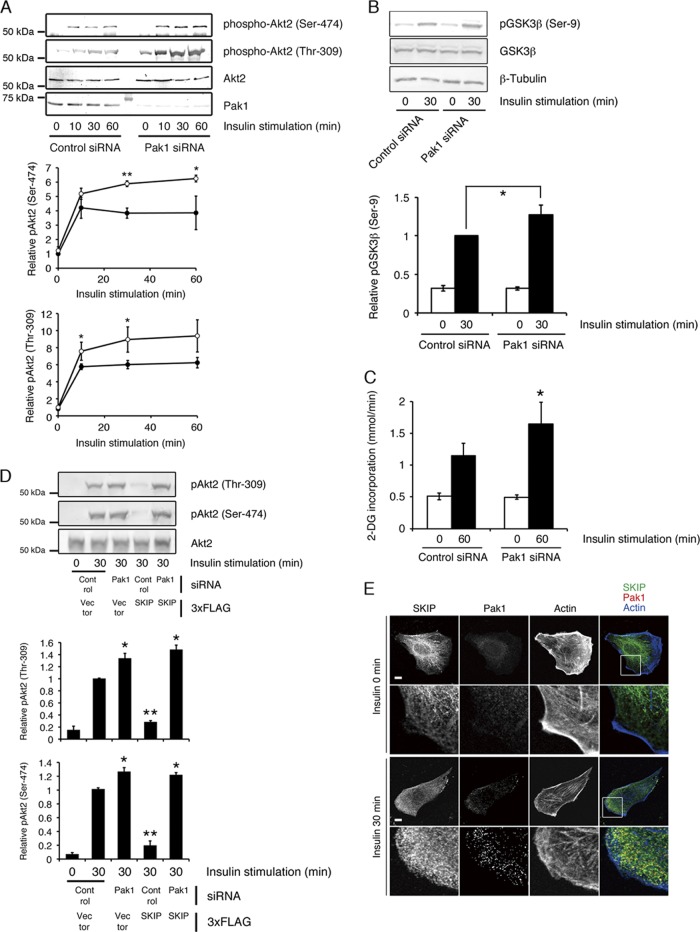
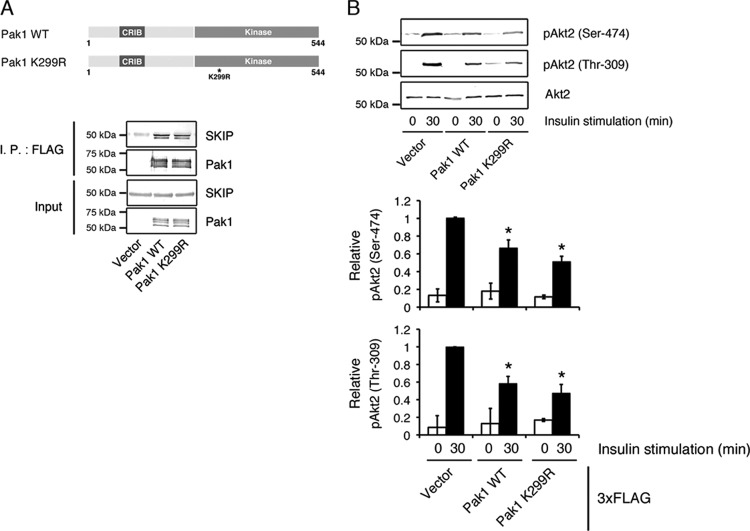
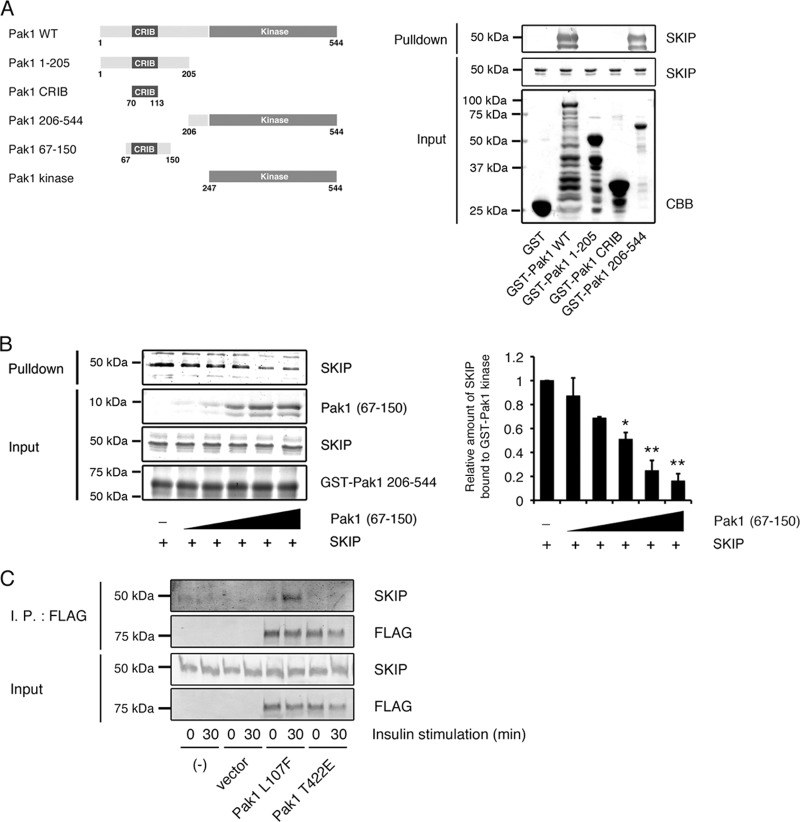
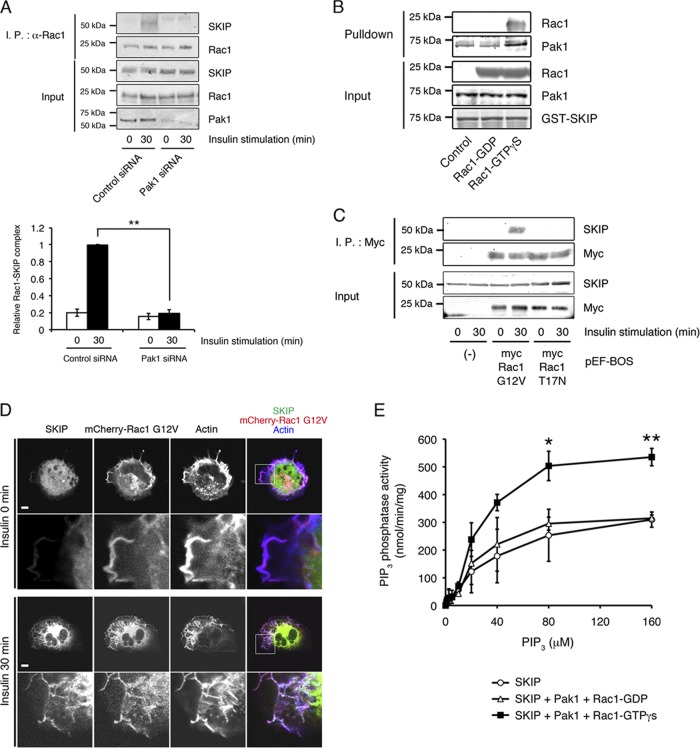
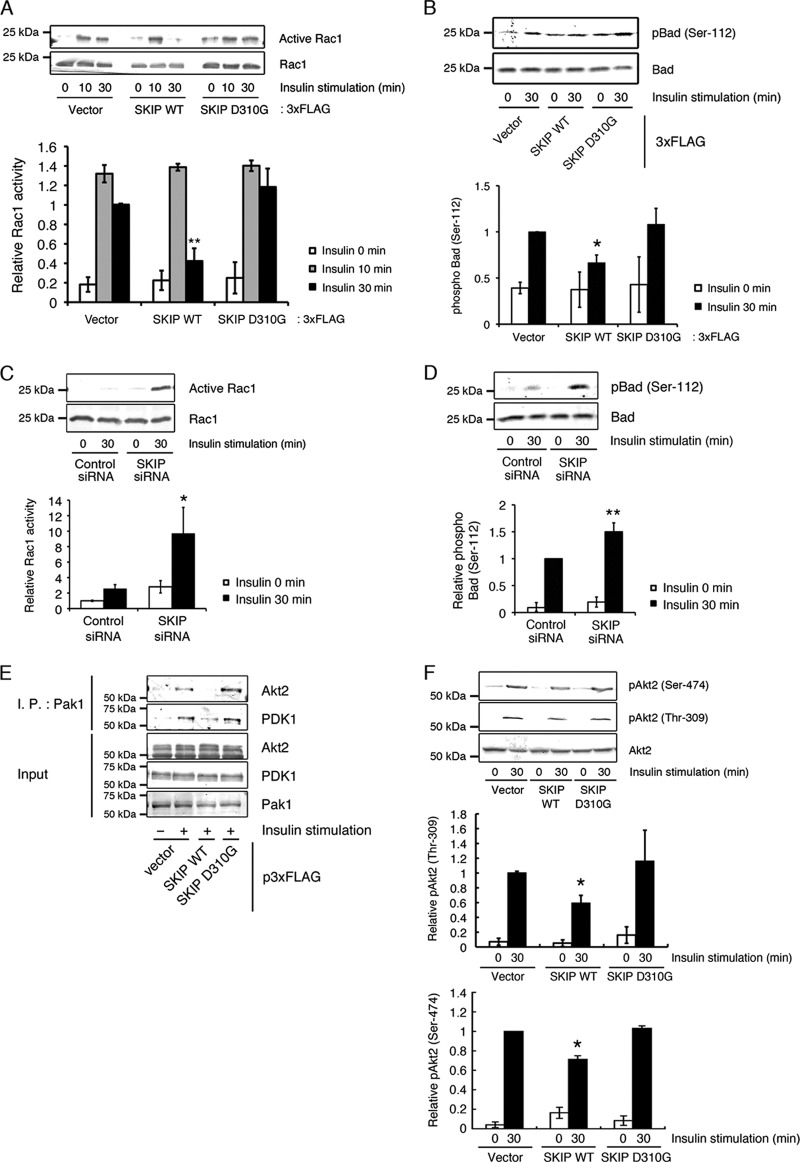
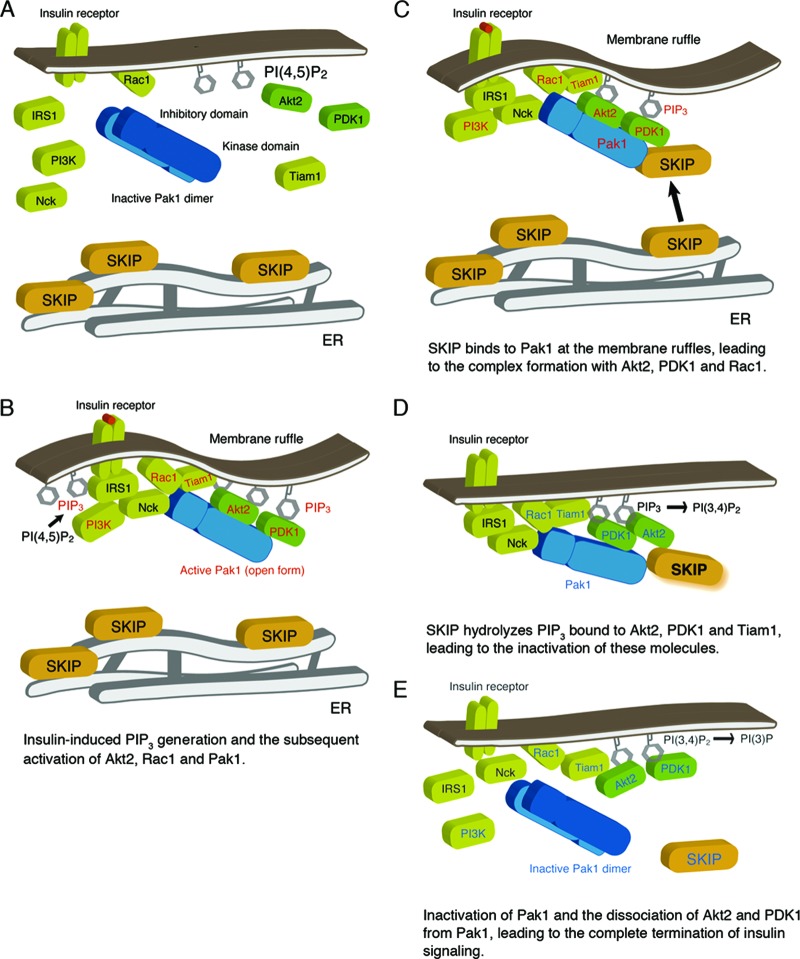
Skeletal muscle and kidney-enriched inositol polyphosphate phosphatase (SKIP) has previously been implicated in the regulation of insulin signaling in skeletal muscle. Here, we present the first report of the mechanisms by which SKIP specifically suppresses insulin signaling and the subsequent glucose uptake. Upon insulin stimulation, SKIP is translocated to the membrane ruffles, where it binds to the active form of Pak1, which mediates multiple protein complex formation with phosphatidylinositol 3,4,5-triphosphate (PIP(3)) effectors such as Akt2, PDK1, and Rac1; this leads to inactivation of these proteins. SKIP also promotes the inhibition of Rac1-dependent kinase activity and the scaffolding function of Pak1, which results in the dissociation of Akt2 and PDK1 from Pak1. Thus, specific suppression of insulin signaling is achieved via the spatiotemporal regulation of SKIP through the scaffolding function of Pak1. These interactions are the foundation of the specific and prominent role of SKIP in the regulation of insulin signaling.
Figures
Similar articles
-
Regulation of insulin signaling and glucose transporter 4 (GLUT4) exocytosis by phosphatidylinositol 3,4,5-trisphosphate (PIP3) phosphatase, skeletal muscle, and kidney enriched inositol polyphosphate phosphatase (SKIP).J Biol Chem. 2012 Mar 2;287(10):6991-9. doi: 10.1074/jbc.M111.335539. Epub 2012 Jan 15. J Biol Chem. 2012. PMID: 22247557 Free PMC article.
-
Improvement of insulin signaling in myoblast cells by an addition of SKIP-binding peptide within Pak1 kinase domain.Biochem Biophys Res Commun. 2015 Jan 2;456(1):41-6. doi: 10.1016/j.bbrc.2014.11.031. Epub 2014 Nov 18. Biochem Biophys Res Commun. 2015. PMID: 25446075
-
Glucose-regulated protein 78 (GRP78) binds directly to PIP3 phosphatase SKIP and determines its localization.Genes Cells. 2016 May;21(5):457-65. doi: 10.1111/gtc.12353. Epub 2016 Mar 4. Genes Cells. 2016. PMID: 26940976
-
Activation of endothelial ras-related C3 botulinum toxin substrate 1 (Rac1) improves post-stroke recovery and angiogenesis via activating Pak1 in mice.Exp Neurol. 2019 Dec;322:113059. doi: 10.1016/j.expneurol.2019.113059. Epub 2019 Sep 6. Exp Neurol. 2019. PMID: 31499064 Free PMC article. Review.
-
Regulation of PtdIns(3,4,5)P3/Akt signalling by inositol polyphosphate 5-phosphatases.Biochem Soc Trans. 2016 Feb;44(1):240-52. doi: 10.1042/BST20150214. Biochem Soc Trans. 2016. PMID: 26862211 Review.
Cited by
-
Atl (atlastin) regulates mTor signaling and autophagy in Drosophila muscle through alteration of the lysosomal network.Autophagy. 2024 Jan;20(1):131-150. doi: 10.1080/15548627.2023.2249794. Epub 2023 Aug 30. Autophagy. 2024. PMID: 37649246 Free PMC article.
-
Defective lysosome reformation during autophagy causes skeletal muscle disease.J Clin Invest. 2021 Jan 4;131(1):e135124. doi: 10.1172/JCI135124. J Clin Invest. 2021. PMID: 33119550 Free PMC article.
-
Rho GTPases-Emerging Regulators of Glucose Homeostasis and Metabolic Health.Cells. 2019 May 9;8(5):434. doi: 10.3390/cells8050434. Cells. 2019. PMID: 31075957 Free PMC article. Review.
-
Phosphatidylinositol 3,4,5-Trisphosphate Phosphatase SKIP Links Endoplasmic Reticulum Stress in Skeletal Muscle to Insulin Resistance.Mol Cell Biol. 2015 Oct 19;36(1):108-18. doi: 10.1128/MCB.00921-15. Print 2016 Jan 1. Mol Cell Biol. 2015. PMID: 26483413 Free PMC article.
-
An Allosteric Binding Site on Sortilin Regulates the Trafficking of VLDL, PCSK9, and LDLR in Hepatocytes.Biochemistry. 2020 Nov 17;59(45):4321-4335. doi: 10.1021/acs.biochem.0c00741. Epub 2020 Nov 5. Biochemistry. 2020. PMID: 33153264 Free PMC article.
References
-
- Carpten JD, et al. 2007. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448: 439– 444 - PubMed
-
- Cho H, et al. 2001. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 292: 1728– 1731 - PubMed
-
- Christoforidis S, Zerial M. 2000. Purification and identification of novel Rab effectors using affinity chromatography. Methods 20: 403– 410 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous