

Cell death and autophagy under oxidative stress: roles of poly(ADP-Ribose) polymerases and Ca(2+)
- PMID: 22751932
- PMCID: PMC3422003
- DOI: 10.1128/MCB.00437-12
Cell death and autophagy under oxidative stress: roles of poly(ADP-Ribose) polymerases and Ca(2+)
Abstract
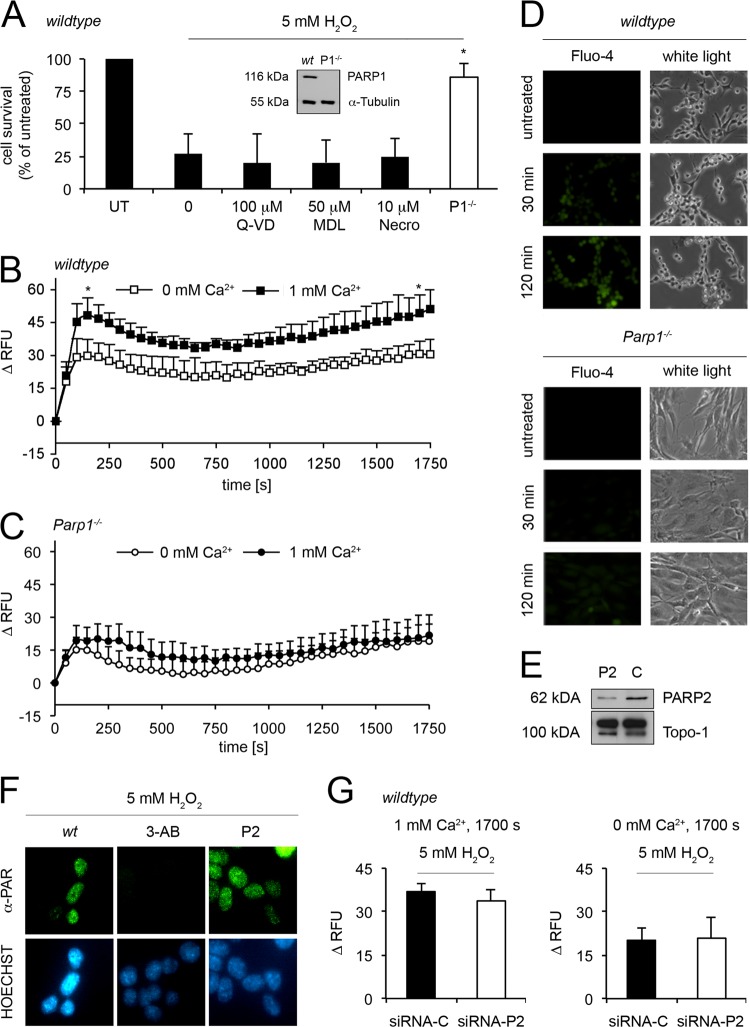
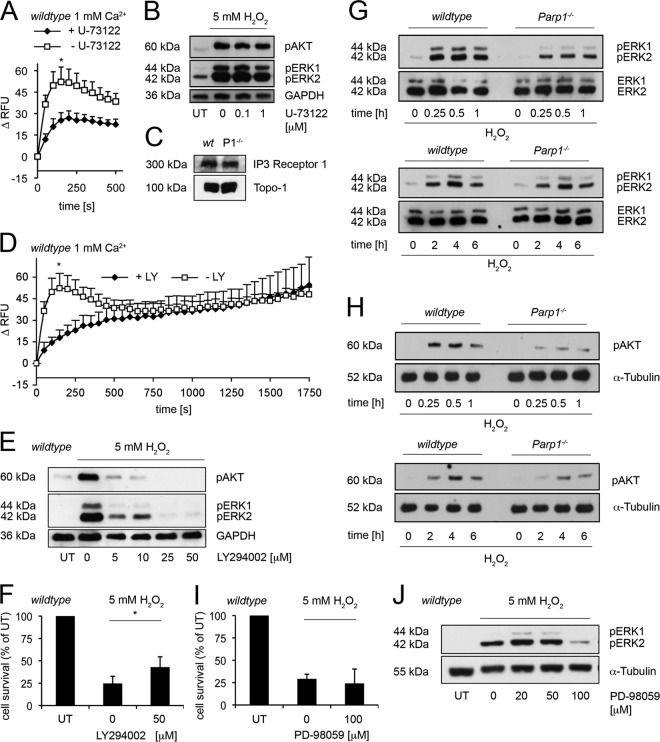
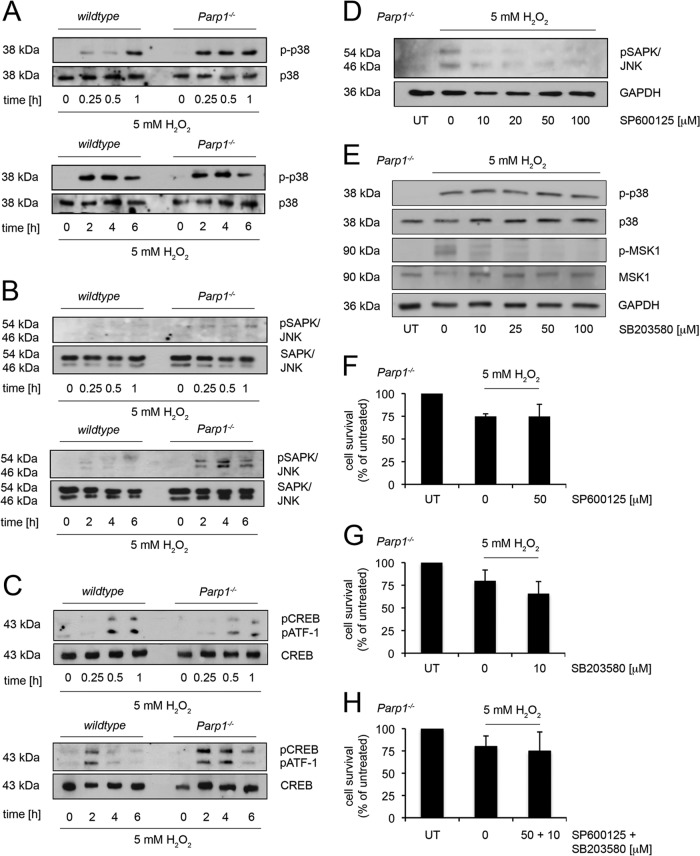
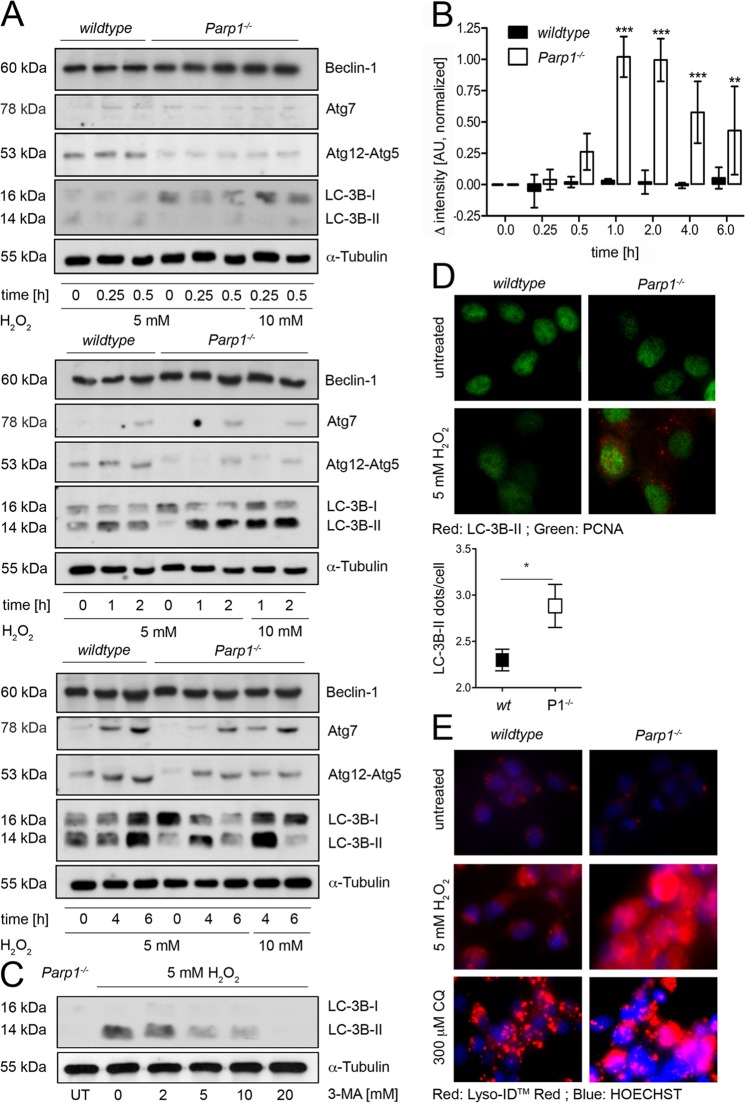
On the cellular level, oxidative stress may cause various responses, including autophagy and cell death. All of these outcomes involve disturbed Ca(2+) signaling. Here we show that the nuclear enzymes poly(ADP-ribose) polymerase 1 (PARP1) and PARP2 control cytosolic Ca(2+) shifts from extracellular and intracellular sources associated with autophagy or cell death. The different Ca(2+) signals arise from the transient receptor potential melastatin 2 (TRPM2) channels located in the cellular and lysosomal membranes. They induce specific stress kinase responses of canonical autophagy and cell death pathways. Autophagy is under the control of PARP1, which operates as an autophagy suppressor after oxidative stress. Cell death is activated downstream of extracellular signal-regulated kinase 1/2 (ERK1/2) and AKT, whereas cell survival correlates with the phosphorylation of p38, stress-activated protein kinase/Jun amino-terminal kinase (SAPK/JNK), and cyclic AMP response element-binding protein (CREB) with its activating transcription factor (ATF-1). Our results highlight an important role for PARP1 and PARP2 in the epigenetic control of cell death and autophagy pathways.
Figures
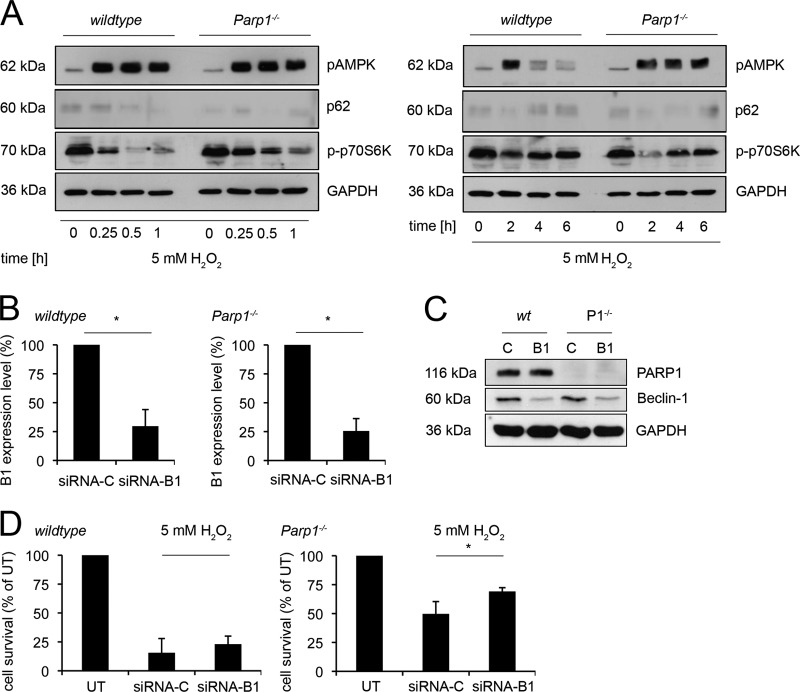
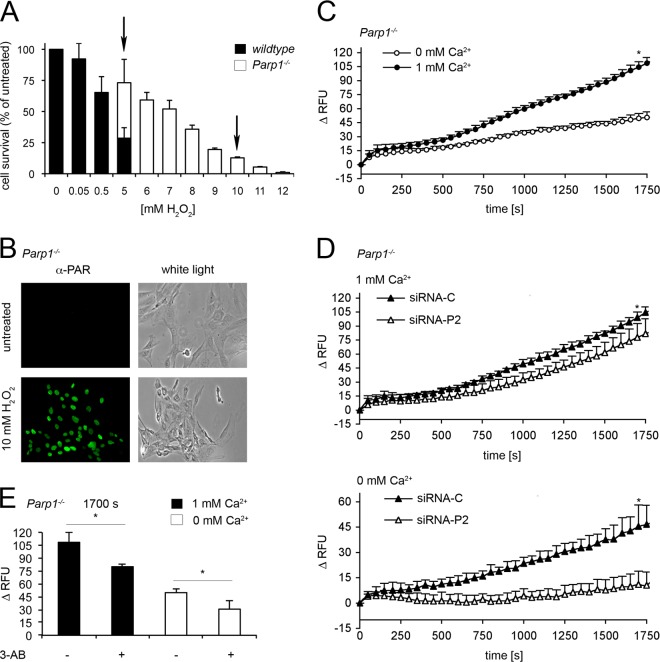
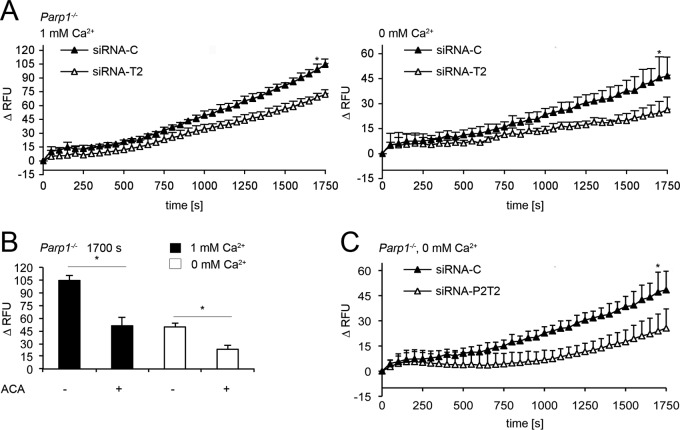

References
-
- Blanc A, Pandey NR, Srivastava AK. 2003. Synchronous activation of ERK 1/2, p38mapk and PKB/Akt signaling by H2O2 in vascular smooth muscle cells: potential involvement in vascular disease (review). Int. J. Mol. Med. 11: 229–234 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous