

Activation of the stress proteome as a mechanism for small molecule therapeutics
- PMID: 22752410
- PMCID: PMC3441123
- DOI: 10.1093/hmg/dds247
Activation of the stress proteome as a mechanism for small molecule therapeutics
Abstract
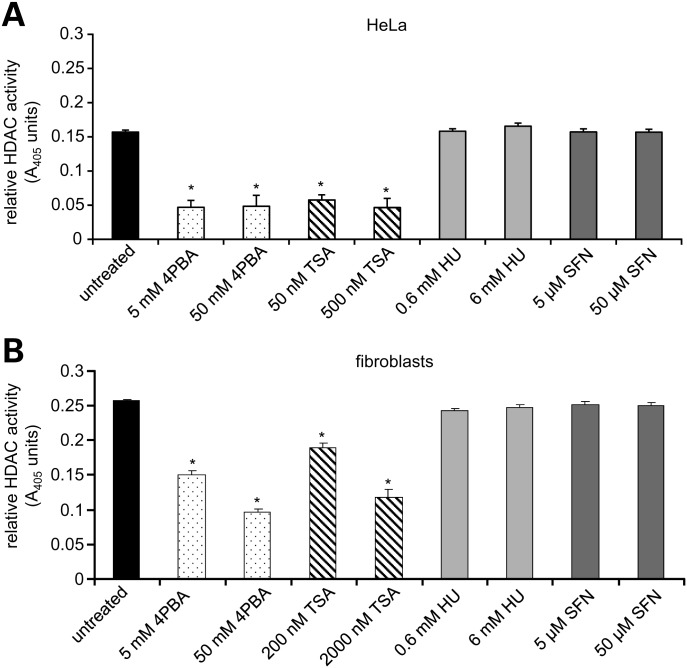
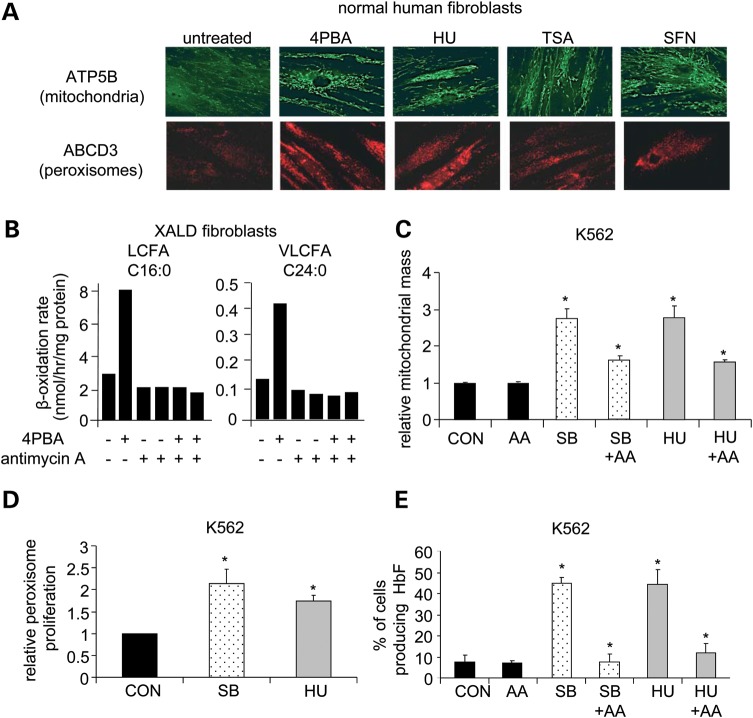
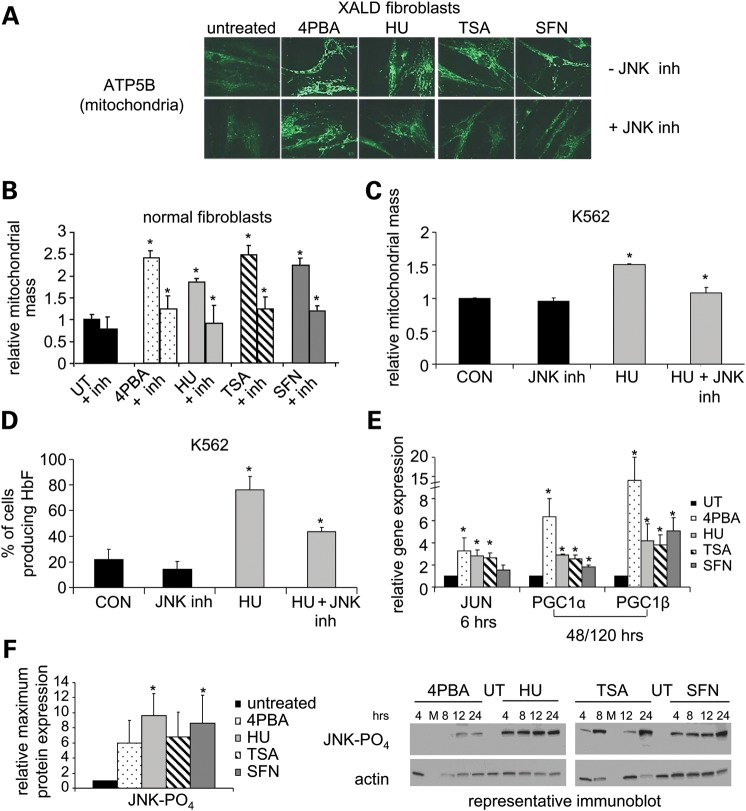
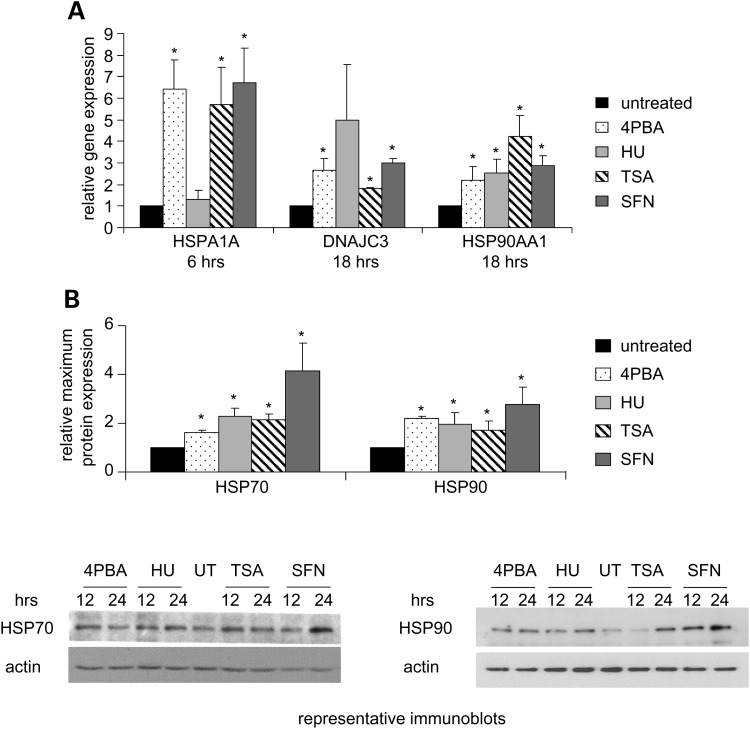
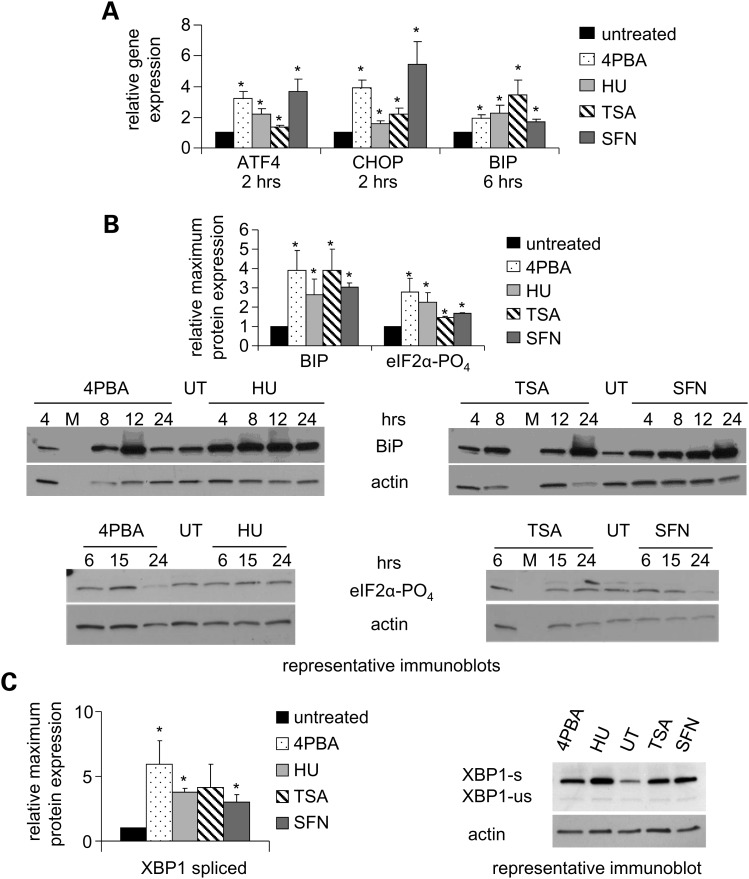
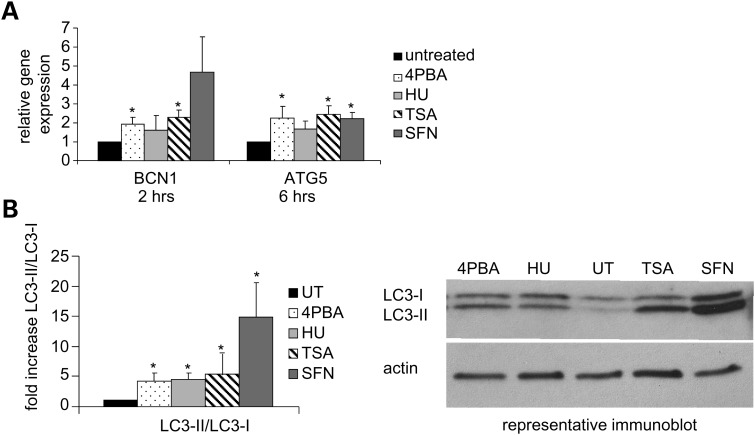
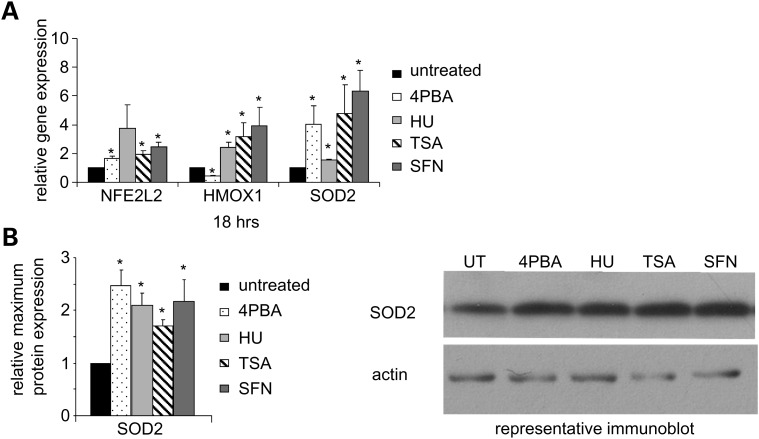
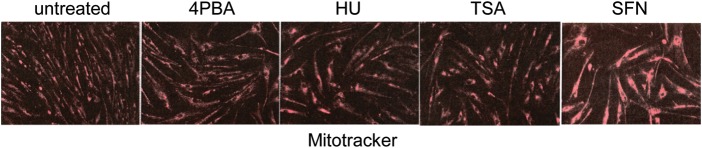
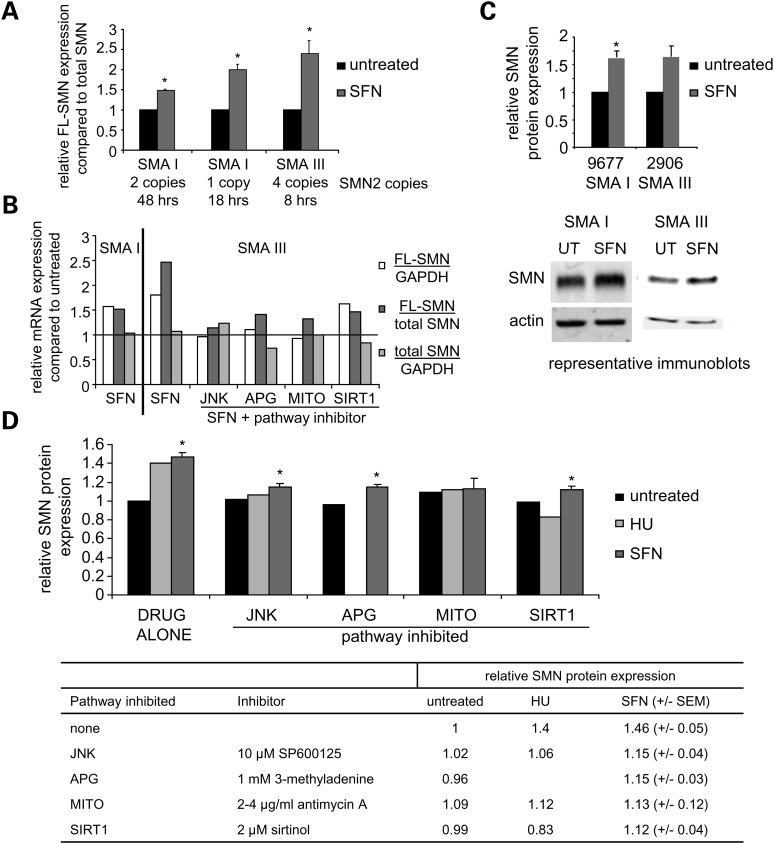
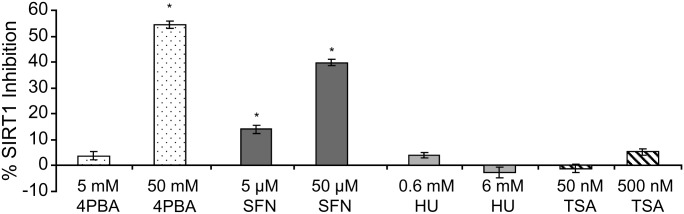
Various small molecule pharmacologic agents with different known functions produce similar outcomes in diverse Mendelian and complex disorders, suggesting that they may induce common cellular effects. These molecules include histone deacetylase inhibitors, 4-phenylbutyrate (4PBA) and trichostatin A, and two small molecules without direct histone deacetylase inhibitor activity, hydroxyurea (HU) and sulforaphane. In some cases, the therapeutic effects of histone deacetylase inhibitors have been attributed to an increase in expression of genes related to the disease-causing gene. However, here we show that the pharmacological induction of mitochondrial biogenesis was necessary for the potentially therapeutic effects of 4PBA or HU in two distinct disease models, X-linked adrenoleukodystrophy and sickle cell disease. We hypothesized that a common cellular response to these four molecules is induction of mitochondrial biogenesis and peroxisome proliferation and activation of the stress proteome, or adaptive cell survival response. Treatment of human fibroblasts with these four agents induced mitochondrial and peroxisomal biogenesis as monitored by flow cytometry, immunofluorescence and/or western analyses. In treated normal human fibroblasts, all four agents induced the adaptive cell survival response: heat shock, unfolded protein, autophagic and antioxidant responses and the c-jun N-terminal kinase pathway, at the transcriptional and translational levels. Thus, activation of the evolutionarily conserved stress proteome and mitochondrial biogenesis may be a common cellular response to such small molecule therapy and a common basis of therapeutic action in various diseases. Modulation of this novel therapeutic target could broaden the range of treatable diseases without directly targeting the causative genetic abnormalities.
Figures
Similar articles
-
Evaluation of pharmacological induction of fatty acid beta-oxidation in X-linked adrenoleukodystrophy.Mol Genet Metab. 2001 Sep-Oct;74(1-2):256-63. doi: 10.1006/mgme.2001.3239. Mol Genet Metab. 2001. PMID: 11592822
-
ABCD1 deletion-induced mitochondrial dysfunction is corrected by SAHA: implication for adrenoleukodystrophy.J Neurochem. 2015 May;133(3):380-96. doi: 10.1111/jnc.12992. Epub 2015 Jan 13. J Neurochem. 2015. PMID: 25393703 Free PMC article.
-
Pharmacological induction of redundant genes for a therapy of X-ALD: phenylbutyrate and other compounds.Adv Exp Med Biol. 2003;544:281-91. doi: 10.1007/978-1-4419-9072-3_36. Adv Exp Med Biol. 2003. PMID: 14713242 Review. No abstract available.
-
Phenylbutyrate up-regulates the adrenoleukodystrophy-related gene as a nonclassical peroxisome proliferator.J Cell Biol. 2005 Apr 11;169(1):93-104. doi: 10.1083/jcb.200501036. Epub 2005 Apr 4. J Cell Biol. 2005. PMID: 15809314 Free PMC article.
-
Modulation of mitochondrial functions by the indirect antioxidant sulforaphane: a seemingly contradictory dual role and an integrative hypothesis.Free Radic Biol Med. 2013 Dec;65:1078-1089. doi: 10.1016/j.freeradbiomed.2013.08.182. Epub 2013 Aug 30. Free Radic Biol Med. 2013. PMID: 23999506 Review.
Cited by
-
Sulforaphane treatment of autism spectrum disorder (ASD).Proc Natl Acad Sci U S A. 2014 Oct 28;111(43):15550-5. doi: 10.1073/pnas.1416940111. Epub 2014 Oct 13. Proc Natl Acad Sci U S A. 2014. PMID: 25313065 Free PMC article. Clinical Trial.
-
(-)-Epicatechin Inhibits Metastatic-Associated Proliferation, Migration, and Invasion of Murine Breast Cancer Cells In Vitro.Molecules. 2023 Aug 24;28(17):6229. doi: 10.3390/molecules28176229. Molecules. 2023. PMID: 37687058 Free PMC article.
-
Leveraging the Mendelian disorders of the epigenetic machinery to systematically map functional epigenetic variation.Elife. 2021 Aug 31;10:e65884. doi: 10.7554/eLife.65884. Elife. 2021. PMID: 34463256 Free PMC article.
-
NRF2 and PPAR-γ Pathways in Oligodendrocyte Progenitors: Focus on ROS Protection, Mitochondrial Biogenesis and Promotion of Cell Differentiation.Int J Mol Sci. 2020 Sep 29;21(19):7216. doi: 10.3390/ijms21197216. Int J Mol Sci. 2020. PMID: 33003644 Free PMC article.
-
Exploring sulforaphane as neurotherapeutic: targeting Nrf2-Keap & Nf-Kb pathway crosstalk in ASD.Metab Brain Dis. 2024 Mar;39(3):373-385. doi: 10.1007/s11011-023-01224-4. Epub 2023 May 30. Metab Brain Dis. 2024. PMID: 37249861 Review.
References
-
- Kemp S., Wei H.M., Lu J.F., Braiterman L.T., McGuinness M.C., Moser A.B., Watkins P.A., Smith K.D. Gene redundancy and pharmacological gene therapy: implications for X-linked adrenoleukodystrophy. Nat. Med. 1998;4:1261–1268. - PubMed
-
- Gardian G., Browne S.E., Choi D.K., Klivenyi P., Gregorio J., Kubilus J.K., Ryu H., Langley B., Ratan R.R., Ferrante R.J., et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington's disease. J. Biol. Chem. 2005;280:556–563. - PubMed
-
- McGuinness M.C., Zhang H.P., Smith K.D. Evaluation of pharmacological induction of fatty acid beta-oxidation in X-linked adrenoleukodystrophy. Mol. Genet. Metab. 2001;74:256–263. - PubMed
-
- Li X., Baumgart E., Dong G.X., Morrell J.C., Jimenez-Sanchez G., Valle D., Smith K.D., Gould S.J. PEX11alpha is required for peroxisome proliferation in response to 4-phenylbutyrate but is dispensable for peroxisome proliferator-activated receptor alpha-mediated peroxisome proliferation. Mol. Cell. Biol. 2002;22:8226–8240. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
